Imagine a world where there are no laws regulating the food you eat. Butchers slaughter cows covered in boils and pile the parts on the floor. Rats are free to crawl about the mountains of meat and organs until they die from eating poisoned bread set out by workers. This mixture of diseased beef parts, dead rats and poison is then shoveled into giant grinders and processed with toxic preservatives to make canned meat. If you’re lucky, the can is labeled «beef,» but it’s just as likely to read «chicken» or «pork» [source: Sinclair].
Now, imagine that medicines are just as questionable. Remedies for kidney and liver problems contain alcohol, making things worse instead of better. Crying or colicky babies are dying because the tonic meant to quiet them contains heavy doses of morphine. But you couldn’t avoid these ingredients if you tried — they aren’t listed on the label [source: McQueeney].
Sounds pretty terrible, right? Even more disturbing, though, is the fact that none of these scenarios is actually made up. This was reality when the scientific, regulatory and public health agency now known as the Food and Drug Administration (FDA) was born in the early 1900s.
From these beginnings, the FDA has grown to regulate more than just food and drugs. Medical devices, electronics that emit radiation, cosmetics, veterinary products, tobacco and biologics (like vaccines and blood) all fall under the agency’s jurisdiction. To monitor these categories, the FDA employed 14,829 total staff as of 2013, many at its Silver Spring, Maryland, headquarters. Its employees come from a wide variety of professions, and they include attorneys, biologists, chemists, engineers, pharmacologists, physicians and statisticians [source: HHS, «About» and FDA, «Career»].
Of course, all these efforts require money — $4.4 billion in 2014, to be exact. Congress and the president authorized $2.6 billion of this funding, while the remaining $1.8 billion came from user fees [source: FDA, «Narrative»]. While this sounds like a lot, it’s actually just a small fraction of the nearly $1 trillion budget of the FDA’s parent agency, the Department of Health and Human Services [source: HHS, «Budget»].
So is this money well-spent? Decide for yourself as you read more stomach-churning stories about pre-FDA life.
Contents
- Agency Origins
- Administration Organization
- A Look at Regulation
- Criticisms and Successes
Agency Origins
They say the easiest way to a man’s heart is through his stomach. Turns out, the same can be said about a politician’s vote. At the turn of the 20th century, numerous revelations about questionable handling and dangerous additives led Congress to pass a food safety law. Known as the Food and Drugs Act, it eventually led to the creation of the FDA, the United States’ oldest federal consumer protection agency.
The origins of the FDA go all the way back to 1862. As the boom of Civil War cannons echoed across the hills of the American South, President Lincoln appointed chemist Charles M. Wetherill to analyze agricultural materials like food, soil and fertilizer in the newly formed Department of Agriculture.
After some two decades of food adulteration investigations, his successors, Peter Collier and Dr. Harvey W. Wiley, became convinced the country needed a law to regulate food and drug contents and quality. Wiley was particularly proactive, going so far as to recruit a group of healthy young men known as «The Poison Squad» to serve as human lab rats for experiments with common, and often poisonous, food additives. Partly because of Wiley’s work, Congress considered more than 100 food and drug bills between 1880 and 1905 [sources: Swann and FDA, «Dates»].
By 1906, however, Congress — heavily influenced by food packing and canning lobbyists — had yet to pass a comprehensive food and drug law. The tipping point was the publication of a novel that was so disgusting, lawmakers had no choice but to act: Upton Sinclair’s «The Jungle.» Though technically fiction, the novel was based on Sinclair’s undercover work in Chicago’s meatpacking plants, and it detailed conditions like the rat-infested mystery meat. One of his most famous claims was that workers who fell into the meat hoppers were sometimes ground in with the rest of the meat!
In response to these shocking allegations, President Theodore Roosevelt ordered an investigation that verified all of what Sinclair had claimed — except for the human hamburger part. On June 30, 1906, Congress passed the Food and Drugs Act (also known as the Wiley Act), prohibiting the trade of mislabeled and contaminated food, beverages and medicines across state lines. Wiley’s Bureau of Chemistry was tasked with enforcing the new law, making it the first real predecessor of the modern FDA [source: Sinclair and FDA, «Dates»].
Administration Organization
Like most government entities, the FDA evolved out of several other agencies and experienced some name changes, reorganizations and relocations before it became the agency we know today. For simplicity’s (and sanity’s) sake, we won’t wade too deeply in the nitty-gritty details. Instead, let’s focus on two simple questions: How was the modern FDA formed, and how is it organized today?
As we learned in the last section, Congress tasked the Department of Agriculture’s Bureau of Chemistry with enforcing the 1906 Food and Drugs Act. To accomplish this task, the agency conducted experiments to inform the regulations they created. In 1927, Congress split these duties between two separate agencies: the Food, Drug and Insecticide Administration (FDIA), which handled the regulations, and the Bureau of Chemistry and Soils, which performed the experiments. Just a few years later, on July 1, 1930, Congress dropped the «insecticide» part and renamed the agency, simply, the Food and Drug Administration. From there, it bounced around under four different parent agencies until 1979, when it finally landed where it is now — under the Department of Health and Human Services [source: National Archives].
The FDA’s organization has certainly changed over its 85-year history, but it’s generally been hierarchical; think layers on a cake. Today, the top layer is the Office of the Commissioner of Food and Drugs, which reports directly to the secretary of Health and Human Services. This first layer also includes seven supporting organizations that report to the commissioner and help the agency with high-level administrative tasks like crafting policy, coordinating outreach and handling litigation [source: FDA, «Commissioner»].
The next layer contains the offices of five deputy commissioners who all answer to the commissioner and do most of the agency’s heavy lifting. Two of these units, the Office of Foods and Veterinary Medicine and the Office of Medical Products and Tobacco, are responsible for ensuring the safety of specific products. The other three — the Office of Global Regulatory Operations and Policy; the Office of Policy, Planning, Legislation and Analysis; and the Office of Operations — support these efforts by offering policy guidance, drafting legislation, assisting with day-to-day operations and providing other general services. Each of these offices has one or more layers below it, all playing a role in keeping our food, drugs and other products safe [source: FDA, «Organization»].
A Look at Regulation
The FDA oversees a staggering amount of goods sold in the United States. According to one estimate, they’re responsible for ensuring the safety of items accounting for 20 cents out of every dollar consumers spend [source: FDA, «Origin»]. Of course, the FDA can’t examine every factory and test every product, so they do the best they can with a mix of regulations, guidelines, inspections and approval processes.
All FDA regulations are governed by more than 100 separate laws passed by Congress. One of the most important is the Food, Drug and Cosmetic Act of 1938, which expanded the 1906 law we discussed earlier. This landmark legislation, among other things, created food standards, allowed for factory inspections and forced drug companies to prove their products are safe. Other milestones include the Kefauver-Harris Amendments of 1962, which greatly bolstered drug safety rules, in addition to the Medical Device Amendments of 1976, which improved the safety of medical devices [source: FDA, «Legislation»].
What, then, is the FDA supposed to regulate? The general categories include foods, drugs, biologics, medical devices, radiation-emitting products, cosmetics, veterinary products and tobacco products. When a regulation is proposed, the FDA accepts public comment before it issues the final rule, which then becomes law. If you’ve got a lot of free time on your hands, you can check them out in Title 21 of the Code of Federal Regulations. Short of a regulation, the FDA can also issue guidance. This is sort of like the assembly instructions for a piece of furniture: It’s the agency’s best advice for how a regulation could be carried out, but it doesn’t necessarily have to be done that way [source: FDA, «Difference»].
To ensure the regulations are being followed, the FDA may inspect some products and facilities. Agency inspectors visit more than 16,000 facilities per year, including food-processing and drug-manufacturing facilities, dairy farms and even blood banks [source: Swann]. Because the FDA can’t examine every facility and product they oversee, they will also conduct impromptu inspections if they receive a specific complaint. Additionally, the agency will approve certain products they’ve reviewed for safety and effectiveness. Depending on the product regulation, this may be done before or after the item goes on the market. Such «FDA-approved» items include drugs and food additives for people and animals, biologics and high-risk medical devices [sources: FDA, «Inspect» and FDA, «Approved»].
Criticisms and Successes
When you turn on the television news these days, there’s a good chance you’ll hear two pundits having a heated debate about the merits of government regulation. Some feel it’s costly and hampers progress while others feel it’s necessary to keep businesses honest. As a regulatory agency, the FDA is certainly no stranger to this controversy. A quick look back at the agency’s history shows that both sides of the regulation debate have some valid points.
One instance when critics felt FDA regulations slowed progress was during the AIDS epidemic of the 1980s. Little was known about the disease, and for many it was a death sentence. Activists felt the FDA was dragging its feet through the approval process for promising new drugs. The conflict came to a head Oct. 11, 1988, when 1,000 protesters managed to close down the agency’s headquarters in Maryland, some holding posters that read, «Time isn’t the only thing the FDA is killing» [source: Crimp].
Others think FDA regulation doesn’t go far enough. These critics point to cases like that of Vioxx, a painkiller approved by the agency in 1999. When a 2000 study showed the drug might pose a significant risk for heart attack when compared to a similar painkiller, Vioxx’s manufacturer, Merck, shrugged it off. The other drug simply had a highly beneficial effect on the heart, they reasoned. The FDA accepted the argument, and Vioxx remained on the market until 2004, when Merck pulled it amid mounting criticism. By this time, according to one study, Vioxx had caused between 88,000 and 140,000 heart attacks [source: Bhattacharya and Berenson et al.].
Still, the FDA has had some undeniable successes, like its refusal to license the sleeping pill thalidomide in the United States. Developed in Germany, scientists initially viewed the drug as exceedingly safe, approving it for over-the-counter use in most European countries by 1956. In 1961, however, scientists discovered a link between thalidomide and children born with malformed limbs. While such birth defects affected 10,000 children worldwide, the United States was largely spared thanks in large part to resistance from the FDA and, specifically, its drug examiner, Frances Oldham Kelsey [source: Science Museum].
Lots More Information
Author’s Note: How the FDA Works
When writing about government agencies like the FDA, things can get dry pretty fast. But hidden beneath snooze-worthy policies, regulations and organizational charts are truly fascinating historical events that influenced the agency’s development in one way or another. There were so many, in fact, that I couldn’t mention them all in the article. There was the story of elixir of sulfanilamide, which killed 107 people, mostly children, and led to the passage of the Federal Food, Drug and Cosmetic Act of 1938. Or the mysterious case of Tylenol capsules laced with cyanide, which led to tamper-resistant packing regulations in 1982. Bureaucracy: It’s more interesting than you think!
Related Articles
- 10 Costly Food Recalls
- Food Labels 101
- How Food Safety Organizations Work
- Labeling Genetically Modified Foods
- Top 10 Weirdest Prescription Drug Side Effects
More Great Links
- Department of Health and Human Services
- U.S. Food and Drug Administration
- Project Gutenberg: The Jungle by Upton Sinclair
Sources
- Berenson, Alex et al. «Despite Warnings, Drug Giant Took Long Path to Vioxx Recall.» The New York Times. Nov. 14, 2004. (March 26, 2015) http://www.nytimes.com/2004/11/14/business/14merck.html?pagewanted=all
- Bhattacharya, Shaoni. «Up to 140,000 heart attacks linked to Vioxx.» Jan. 25, 2005. (March 26, 2015) http://www.newscientist.com/article/dn6918-up-to-140000-heart-attacks-linked-to-vioxx.html
- Crimp, Douglas. «Before Occupy: How AIDS Activists Seized Control of the FDA in 1988.» The Atlantic. Dec. 6, 2011. (March 26, 2015) http://www.theatlantic.com/health/archive/2011/12/before-occupy-how-aids-activists-seized-control-of-the-fda-in-1988/249302/
- Janssen, Wallace. «The Story of the Laws Behind the Labels.» FDA Consumer. June 1981. http://www.fda.gov/AboutFDA/WhatWeDo/History/Overviews/ucm056044.htm
- McQueeney, Kerry. «Cocaine for Toothache, Morphine for Your Child’s Cough: The Bizarre ‘Safe Cures’ of 19th Century that ‘Work like Magic.'» Daily Mail. Sept. 4, 2012. (March 24, 2015) http://www.dailymail.co.uk/news/article-2198086/Victorian-adverts-health-remedies-laden-cocaine-morphine-alcohol.html
- Science Museum. «Thalidomide.» (March 26, 2015) http://www.sciencemuseum.org.uk/broughttolife/themes/controversies/thalidomide.aspx
- Sinclair, Upton. «The Jungle: Centennial Edition.» Modern Library Paperbacks. June 6, 2006. http://www.amazon.com/Jungle-Modern-Library-Paperbacks/dp/0812976231/
- Swann, John P. «FDA’s Origin.» U.S. Food and Drug Administration. 1998. http://www.fda.gov/AboutFDA/WhatWeDo/History/Origin/ucm124403.htm
- U.S. Department of Health & Human Services. «About HHS.» Oct. 6, 2014. (March 23, 2015) http://www.hhs.gov/about/
- U.S. Department of Health & Human Services. «Fiscal Year 2014 Budget in Brief: Strengthening Health and Opportunity for All Americans.» March 12, 2014. (March 23, 2015) http://www.hhs.gov/budget/fy2014/fy-2014-budget-in-brief.pdf
- U.S. Food and Drug Administration. «About the Office of Regulatory Affairs.» March 4, 2015. (March 31, 2015) http://www.fda.gov/AboutFDA/CentersOffices/OfficeofGlobalRegulatoryOperationsandPolicy/ORA/default.htm
- U.S. Food and Drug Administration. «About the Office of the Commissioner.» Feb. 25, 2015. (March 25, 2015) http://www.fda.gov/AboutFDA/CentersOffices/OC/default.htm
- U.S. Food and Drug Administration. «Career Descriptions.» Oct. 19, 2011. (March 31, 2015) http://www.fda.gov/AboutFDA/WorkingatFDA/CareerDescriptions/default.htm
- U.S. Food and Drug Administration. «FDA Organization.» Feb. 25, 2015. (March 26, 2015) http://www.fda.gov/AboutFDA/CentersOffices/default.htm
- U.S. Food and Drug Administration. «FY 2014 Operating Plan Narrative.» (March 24, 2015) http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/BudgetReports/UCM388299.pdf
- U.S. Food and Drug Administration. «Harvey W. Wiley.» January — February 2006. (March 25, 2015) http://www.fda.gov/AboutFDA/WhatWeDo/History/CentennialofFDA/HarveyW.Wiley/
- U.S. Food and Drug Administration. «History.» March 23, 2015. (March 24, 2015) http://www.fda.gov/AboutFDA/WhatWeDo/History/
- U.S. Food and Drug Administration. «Is it Really FDA Approved?» Sept. 30, 2008. (March 27, 2015) http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm047470.htm
- U.S. Food and Drug Administration. «Legislation.» March 11, 2015. (March 26, 2015) http://www.fda.gov/RegulatoryInformation/Legislation/default.htm
- U.S. Food and Drug Administration. «ORA Overview.» Aug. 12, 2014. (March 31, 2015) http://www.fda.gov/AboutFDA/CentersOffices/OfficeofGlobalRegulatoryOperationsandPolicy/ORA/ucm409371.htm
- U.S. Food and Drug Administration. «Significant Dates in U.S. Food and Drug Law History.» Dec. 19, 2014. (March 26, 2015) http://www.fda.gov/AboutFDA/WhatWeDo/History/Milestones/ucm128305.htm
- U.S. Food and Drug Administration. «What Does FDA Inspect?» Nov. 18, 2014. (March 26, 2015) http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194888.htm
- U.S. Food and Drug Administration. «What Does FDA Regulate?» Nov. 18, 2014. (March 25, 2015) http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194879.htm
- U.S. Food and Drug Administration. «What is the Difference Between the Federal Food, Drug and Cosmetic Act, FDA Regulations, and FDA Guidance?» Nov. 18, 2014. (March 26, 2015) http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194909.htm
- U.S. Food and Drug Administration. «When and Why Was FDA Formed?» Nov. 18, 2014. (March 23, 2015) http://www.fda.gov/aboutfda/transparency/basics/ucm214403.htm
- U.S. National Archives and Records Administration. «Records of the Food and Drug Administration.» (March 25, 2015) http://www.archives.gov/research/guide-fed-records/groups/088.html
Многие производители хотели бы поставлять свою продукцию на рынок США, ведь там достаточно платежная аудитория и предполагаемая выручка сможет вырасти в разы. Но практически по первому же запросу в Google вы можете узнать, что основным препятствием для выхода на американский рынок является получение разрешения от FDA.
В сегодняшней статье поговорим о том, что же такое FDA, что она контролирует и рассмотрим несколько конкретных продуктов, которые “одобряет” или “не одобряет” FDA.
Что такое FDA и как она работает?
FDA (Food and Drug Administration) или Управление по санитарному надзору за качеством пищевых продуктов и медикаментов — это агентство Министерства здравоохранения и социальных служб США, один из федеральных исполнительных департаментов.
Управление занимается контролем качества пищевых продуктов, лекарственных препаратов, косметических средств, табачных изделий и некоторых других категорий товаров, а также осуществляет контроль за соблюдением законодательства и стандартов в этой области.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов — старейшее агентство по комплексной защите прав потребителей в США. Несмотря на то, что до 1930 года оно не было известно под своим нынешним названием, современные регулирующие функции FDA начались с принятия Закона о чистых пищевых продуктах и лекарствах 1906 года. Закон запрещал межгосударственную торговлю фальсифицированными продуктами питания и лекарствами с неправильным брендом.
С тех пор FDA изменилось вместе с социальными, экономическими, политическими и правовыми изменениями в Соединенных Штатах. Изучение истории этих изменений проливает свет на меняющуюся роль, которую FDA играет в укреплении общественного здоровья, и предлагает уроки, которые следует учитывать при оценке текущих нормативных проблем.
Своей миссией управление по санитарному надзору за качеством пищевых продуктов и медикаментов несет называет:
-
ответственность за охрану здоровья населения,
-
обеспечение безопасности и эффективности лекарственных и ветеринарных препаратов, биологических продуктов и медицинских устройств,
-
обеспечение безопасности продуктов питания и косметики.
FDA также несет ответственность за регулирование производства, маркетинга и распределения табачных изделий с целью защиты здоровья населения и сокращения употребления табака несовершеннолетними.
FDA отвечает за улучшение здоровья населения, помогая ускорить внедрение инноваций для большей эффективности, безопасности и доступности медицинских продуктов.
Что регулирует FDA?
Объем регулирующих полномочий FDA очень широк. Ее обязанности тесно связаны с обязанностями нескольких других правительственных агентств. Производителей часто разочаровывает и сбивает с толку выбор соответствующего регулирующего органа, к которому следует обратиться. Ниже список традиционно признанных категорий продуктов, подпадающих под юрисдикцию FDA.
В целом FDA регулирует:
-
Продукты питания, в том числе:
-
пищевые добавки;
-
бутилированная вода;
-
пищевые добавки;
-
детские смеси;
-
другие продукты питания (хотя Министерство сельского хозяйства США играет ведущую роль в регулировании некоторых продуктов из мяса, птицы и яиц).
-
Лекарства, в том числе:
-
рецептурные препараты (как фирменные, так и дженерики);
-
лекарства, отпускаемые без рецепта (без рецепта).
-
Биопрепараты, в том числе:
-
вакцины для человека;
-
кровь и продукты крови;
-
продукты клеточной и генной терапии;
-
ткани и салфетки;
-
аллергены.
-
Медицинское оборудование, в том числе:
-
простые предметы;
-
сложные технологии, такие как кардиостимуляторы;
-
стоматологические устройства;
-
хирургические имплантаты и протезы.
-
Электронные изделия, испускающие радиацию, в том числе:
-
микроволновые печи;
-
рентгеновское оборудование;
-
лазерная продукция;
-
оборудование для ультразвуковой терапии;
-
ртутные лампы;
-
солнечные лампы.
-
Косметика, в том числе:
-
цветные добавки, содержащиеся в косметике и других средствах личной гигиены;
-
увлажняющие и очищающие средства для кожи;
-
лак для ногтей;
-
духи.
-
Ветеринарные продукты, в том числе:
-
корма для скота;
-
корма для домашних животных;
-
ветеринарные препараты и устройства.
-
Табачные изделия.
Под контролем FDA находится целый ряд агентств, которые регулируют тот или иной продукт, попадающий под проверку FDA. Среди них:
-
Центр экспертизы и изучения биологических препаратов (Center for Biologics Evaluation and Research);
-
Центр по контролю оборудования и радиационной безопасности (Center for Devices and Radiological Health);
-
Центр оценки и исследований лекарственных средств (Center for Drug Evaluation and Research);
-
Центр безопасности пищевых продуктов и практическим вопросам питания (Center for Food Safety and Applied Nutrition);
-
Центр контроля табачных изделий (Center for Tobacco Products);
-
Центр ветеринарной медицины (Center for Veterinary Medicine);
-
Национальный центр токсикологических исследований (National Center for Toxicological Research);
-
Следственное управление (Office of Criminal Investigations);
-
Бюро обеспечения соответствия нормативным требованиям (Office of Regulatory Affairs).
Что значит “одобрено FDA”?
Возможно, вы видели эти слова на веб-сайте компании или в рекламе какого-либо продукта. Некоторые маркетологи могут сказать, что их продукция «одобрена FDA», но как узнать наверняка, что одобряет Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США?
FDA несет ответственность за охрану здоровья населения, регулируя использование лекарственных и биопрепаратов для людей, лекарственных препаратов для животных, медицинских устройств, табачных изделий, продуктов питания (включая продукты питания для животных), косметики и электронных продуктов, излучающих радиацию.
Но не все эти продукты проходят предварительное одобрение, то есть проверку безопасности и эффективности экспертами FDA и одобрение агентства перед продажей продукта. В некоторых случаях FDA может проверять продукты после того, как они уже выставлены на продажу.
FDA не одобряет компании.
FDA не «одобряет» медицинские учреждения, лаборатории или производителей. FDA имеет право проверять эти объекты, чтобы убедиться, что они соответствуют применимым нормам надлежащей практики.
FDA одобряет новые лекарства и биопрепараты.
Новые лекарства и определенные биопрепараты должны подтверждать свою безопасность и эффективность согласно требованиям FDA до начала продажи. К ним относятся:
-
терапевтические белки,
-
вакцины,
-
кровь и продукты крови.
Производители также должны доказать, что они могут производить лекарственный продукт в соответствии с федеральными стандартами качества.
Эксперты FDA рассматривают результаты лабораторных испытаний, клинических испытаний на животных и людях, проведенных производителями. Если FDA дает разрешение,значит преимущества продукта перевешивают известные риски для предполагаемого использования и продукция может продаваться на внутреннем рынке США.
FDA не одобряет табачные изделия.
Безопасного табачного изделия не существует, поэтому безопасный и эффективный стандарт FDA для оценки медицинских изделий не подходит для табачных изделий. Вместо этого FDA регулирует табачные изделия на основе стандарта общественного здравоохранения, который учитывает риски, связанные с табачными изделиями для населения в целом.
FDA одобряет пищевые добавки для людей.
Хотя FDA не имеет допуска на рынок пищевых продуктов, оно имеет право утверждать определенные ингредиенты до их использования в пищевых продуктах. К ним относятся пищевые добавки, например вещества, намеренно добавленные в пищу, и красители.
Компании, которые хотят добавлять в пищу новые пищевые добавки, несут ответственность за предоставление FDA информации, демонстрирующей безопасность этих добавок. Эксперты FDA проверяют результаты соответствующих тестов, чтобы убедиться, что пищевая добавка безопасна для использования. Одобренная пищевая добавка должна использоваться в соответствии с утвержденными областями применения, спецификациями и ограничениями.
Также FDA должно одобрить красители, которые используются в пище (включая корм для животных), диетических добавках, лекарствах, косметике и некоторых медицинских устройствах. Эти красящие добавки (за исключением красок для волос на основе каменноугольной смолы) подлежат утверждению агентством, и каждая из них должна использоваться только в соответствии с утвержденными областями применения, спецификациями и ограничениями.
FDA одобряет лекарства для животных и пищевые добавки для использования в кормах для животных.
FDA отвечает за одобрение лекарств для животных, включая домашних животных, домашний скот и птицу. Хотя FDA не разрешает маркетинг кормов для животных, они одобряют пищевые добавки, используемые в этих продуктах.
FDA работает над тем, чтобы гарантировать, что корм для животных безопасен, изготовлен в санитарных условиях и должным образом маркирован.
Правило превентивного контроля пищевых продуктов для животных, утвержденное Законом о модернизации безопасности пищевых продуктов FDA (FSMA), требует, чтобы пищевые компании принимали меры для предотвращения загрязнения пищевых продуктов и использовали текущие надлежащие производственные практики (такие как гигиенические методы работы персонала, адекватные санитарные правила и правильное использование оборудования) при приготовлении корма для животных
FDA не одобряет косметику.
Примерами косметических средств являются духи, продукты для макияжа увлажняющие средства, шампуни, краски для волос, очищающие средства для лица и тела, а также средства для бритья. Косметические продукты и ингредиенты, а также их маркировка не требуют одобрения FDA перед тем, как поступить на рынок. Косметика должна быть безопасной для использования по назначению и иметь соответствующую маркировку.
FDA не одобряет продукты медицинского назначения.
Лечебное питание используется для диетического лечения заболевания или состояния здоровья, которое требует особых питательных веществ. Примером лечебного питания является пища для людей с фенилкетонурией, генетическим заболеванием. Человеку с этим заболеванием могут потребоваться лечебные продукты, в состав которых не входит фенилаланин. Лечебное питание предназначено для употребления под наблюдением врача. В него не входят такие продукты, как заменители еды или диетические коктейли, а также продукты для лечения таких заболеваний, как диабет, с которыми можно справиться путем изменения обычной диеты.
Медицинские продукты не должны проходить предварительное одобрение FDA. Но компании по производству медицинских продуктов питания должны соблюдать и другие требования, такие как надлежащая производственная практика и регистрация предприятий питания. Любые заявления в маркировке медицинских пищевых продуктов должны быть правдивыми и не вводить в заблуждение.
FDA не одобряет детские смеси.
FDA не одобряет детские смеси до того, как они появятся на рынке. Но производители детских смесей подлежат регулирующему надзору FDA. Производители должны обеспечить соответствие детской смеси федеральным требованиям к питательным веществам. Производители должны зарегистрироваться в FDA и предоставить агентству уведомление, прежде чем продавать новую формулу.
FDA проводит ежегодные проверки всех предприятий, производящих детские смеси, а также собирает и анализирует образцы продукции. FDA также проверяет новые объекты. Если FDA определяет, что детская смесь представляет риск для здоровья человека, производитель смеси должен провести отзыв продукции с рынка.
Итоги
Стоит иметь в виду, что логотип FDA предназначен только для официального правительственного использования. Логотип FDA не должен использоваться для искажения информации об агентстве или для утверждения, что FDA одобрило какую-либо частную организацию, продукт или услугу в маркетинговых целях.
Таким образом, под обязательную проверку FDA перед началом продаж попадает достаточно большое количество продукции разных отраслей. И даже если ваша продукция не попадает под обязательную проверку, необходимо удостовериться, что ее компоненты не подлежат обязательной предварительной проверке. Достаточно четких и понятных инструкций по этому вопросу есть непосредственно на сайте fda.gov, а если вам что-то в них непонятно, вы можете отправить им письмо для получения разъяснений.
Также вы можете обратиться в нашу компанию и мы поможем Вам с получением необходимых разрешений для поставок в США пищевой и косметической продукции.
Today’s guide explores everything you need to know about FDA.
So, if you have any question about FDA, you will find the answer right here.
Keep reading to learn more.
- What Is FDA?
- What Is The Main Purpose Of The FDA?
- What Products Are Subject To FDA?
- What Are FDA Entry Types?
- What Is The FDA Entry Process?
- What Does It Mean For A Product To Be FDA Approved?
- What Is The Importance Of FDA Approval In Shipping?
- Do I Need To Register With The FDA?
- What Products Do Not Need FDA Approval?
- How Does The FDA Classify Medical Products?
- What Is A Premarket Approval In Relation To FDA Regulations?
- When Is A Premarket Approval Required When Shipping FDA Regulated Products?
- What Is The Difference Between Premarket Approval And 510K Premarket Notification?
- How Long Does FDA Approval Take?
- What Is The Difference Between “FDA Registered” And “FDA Approved” And “FDA Cleared”?
- What Is An FDA Hold In Shipping?
- Why Would The FDA Hold A Shipment?
- How Long Can The FDA Hold A Package?
- What Happens When The FDA Holds A Shipment?
- How Do I Perform An FDA Hold Status Check?
- What Can I Do If My Package Is Held By The FDA?
- What Is The FDA Product Code?
- What Is The Process For Importing FDA Regulated Products?
- Can A Customs Broker Help With FDA Custom Clearance?
- What Is The Cost Of Importing FDA Regulated Products?
What Is FDA?

food and drug administration
We can define food and drug administration as an agent that oversees the process of production of different products that we use and consume in our day-to-day lives.
The food and drug administration monitors products such as cosmetic products, medical equipment, biological products, and food.
They also regulate products that present to be a hazard. That is an emission of radiation which is harmful to the consumer of the products.
What Is The Main Purpose Of The FDA?
Food and drug administration’s perform a wide role in regulating what products the end-user purchases and consumes.
The following are some of the benefits of food and drug administration:
- Facilitate advancing of public health by ensuring a speed up process during product innovation.
- FDA offers protection of the public consumer from products that emit radiation for instance-rays
- They also offer regulations on the amount of content on tobacco products
- They inspect all the cosmetic products and food to ensure proper labeling to prevent consumers from using the wrong product.
This will cause effects on the body. - They also ensure that all medical devices, vaccines, and biological products that humans use are effective and safe for them.
What Products Are Subject To FDA?
The food and drug administration regulates consumer products to protect public health.
There are certain products that FDA highly regulates.
These products include:
- Medical products: such products include, first aid kits, surgical instruments among others. The food and drug administration inspects the standards of these products to prevent preventing under quality equipment.
This will pose a threat to public health. - Human food: FDA regulates what consumers take in terms of food, for instance, excess intake of food additives will cause health issues to the consumer.
- Another case is the dietary supplements that cause an increase in weight when the consumer takes them.
- This will lead to accumulations of fats in the body which is dangerous to your health.
- Human drugs: regulates and inspects all drugs coming in from other countries.
This prevents an increase of counterfeit drugs which will cause a serious problem to the consumer - Cosmetic products: inspects these products to evaluate if the ingredients used meets the threshold.
As a result, preventing any side effects on the consumer’s body and face.
These products include face creams, shampoo, artificial eyelashes among others. - Radiation emitting electronic products such as microwaves, X-ray equipment, LEDs, Laser printers pose a health crisis to the public. users.
The FDA ensures low emitting devices are available with guidelines of their usage. - Tobacco products: FDA ensures the percentage of tobacco in cigarettes, e-cigarettes, and smokeless tobacco meets the specifications given threshold.
- High nicotine in these products causes health issues among the users such as lung cancer due to the high concentration of the substance.
- Biological products, vaccines, and blood: FDA ensures accurate examination of these products before the consumer receives them.
For instance, you should examine the blood for the presence of additional elements, also the body organs should be a match and to the receiver to avoid health crises.
What Are FDA Entry Types?

FDA entry types
Entry type refers to all types of goods that a country allows in via the help of regulatory bodies like the FDA and CPB.
FDA entry uses an Automated broker interface program to achieve the entries electronically.
Food and drug administration process different types of entries. These FDA entries include
Consumption Entry: allows importing goods into the US without issuing requests for the goods and the time.
You can either use these goods for commercial, personal, or business purposes.
With consumption entry, different individuals are responsible for this entry.
They include:
- The owner
- Custom brokers
- Purchaser
We can further classify consumption entry into:
- Informal entry
- Formal entry
In cases of warehouse and import for export FDA entries, they help in removing goods that are entering us.
Other FDA entries include;
- Immediate transportation
- Transportation and exportation
- Temporary importation under bond
- Foreign trade zone
What Is The FDA Entry Process?
The food and Drug Administration entry process involves the steps that custom brokers follow while shipping the products into the country.
These processes include:
If these products are FDA regulated, the customs broker submits custom entry documents to the FDA.
In other cases, the customs broker will submit prior notice to the FDA in case they need more time.
Usually, the customs broker should submit this notice two hours before the arrival of the shipment.
This gives the FDA more time to pre-examine all entries and decide on whether the shipment will cross or needs more examination.
We recommend custom brokers to ensure that products enter the country during the day to allow reviewing at the arrival port.
In case the FDA issues reviews of the products, custom brokers will send a notification to the owner to hold goods longer.
This allows the customs broker to clear any pending issues with FDA to prevent additional charges or refusal of goods entry.
What Does It Mean For A Product To Be FDA Approved?
The main responsibility of food and drug administration is to carry out tests, examinations, reviews and provide approval of products for human consumption.
These products range from medical devices, food, cosmetics, tobacco products, and biological products.
Therefore, for FDA to approve these products they must meet a certain threshold which means they do not pose any danger to the consumers.
Hence, protecting the consumers’ health.
What Is The Importance Of FDA Approval In Shipping?
Food and Drug Administration is important in shipping in the following ways:
- Counterchecks all paper documents to ensure that the shipment does not pose a danger to public health.
- They implement final rules on FDA regulated products such as biological products, food, and medical devices.
- It reduces the holding of goods at the port when the owner provides relevant documents.
- FDA approval also fastens the process of shipping.
This means if the products qualify and adhere to the law FDA will release them immediately.
Do I Need To Register With The FDA?
Yes. To allow easy importation of these products it is important to register with food and drug administration.
Normally, when importing these products into us, you need to register because each product has specific registration variations.
Below are some examples of products and how to register them;
Cosmetic products you should check in with voluntary cosmetic registration program which works as FDA post market reporting system.
This body provides approvals of cosmetic products for manufacturers and distributors.
Tobacco products: if your business in the production, selling of this product you should register with food and drug administration.
You should include sources and types of materials you are using to manufacture these products.
What Products Do Not Need FDA Approval?

Products that do not need FDA approval
In some cases, the food and drug administration does not approve all the products the importers are shipping into the country.
Some of these products pose health risks to the consumer and therefore the FDA will take a longer time to review this product.
These products that do not require food and drug administration approval include;
Tobacco Products
We all know what danger tobacco products pose to human health.
Therefore, the FDA standards examining medical products can not apply to tobacco products.
The law requires you to obtain a written food and drug administration order to allow the production and selling of tobacco products.
The dangers of tobacco products will always bring setbacks to the manufacturer while approaching the market.
As a result, the manufacturer can use the following ways to penetrate the market:
- Premarket tobacco applications
- Substantial equivalence applications
- Exemptions from equivalence applications.
- Compounded drugs
They are a result of a combination of different elements by the pharmacies or doctors to produce medication that best suits patient needs.
FDA approved medicines contain elements that may cause an allergic reaction to different patients when they take these medicines.
Therefore, the Food and Drug Administration does not approve these compound drugs due to the health issue they pose to patients.
Cosmetic Products
As the manufacturer of these products, it’s your responsibility to accurately label and give a description of their usage to consumers.
The food and drug administration does not need to approve these products before reaching the market.
This is because manufacturers will always find ways of penetrating these products to the market.
Infant Formula
The marketing of infant formula allows consumers to sample these products. Thereafter the FDA will evaluate consumer feedback on the infant formula
If this product poses a threat to public health, the FDA declines to approve this product.
In cases of a new brand of infant formula, the manufacturer should provide samples and ingredients for examination to the FDA authorities.
Medical Foods
Normally, we use medical foods to care for the needs of the patients who require specific nutrients foods.
For instance, patients who are diabetic require insulin to low the sugars in the body
Additionally, the FDA does not require the manufacturer to label the products but rather provides accurate information.
How Does The FDA Classify Medical Products?
We can classify medical products into three classes.
It is important to acquire relevant information that helps you understand how these classes work. This normally occurs before submission to the food and drug administration.
This classes results from 16 medical specialists.
These classes include:
Class 1
This class contains general controls providing exemption and in some cases no exemptions
We do not associate this device with high risks of danger
Examples of class1 medical devices are, face masks, bandages,
Class 1 with exemption devices requires the 510k for marketing purposes
Class 2
This class contains the general controls and special controls providing exemption or no exemptions.
This class also requires 510k for a device with an exemption.
Examples; wheelchairs
Class 3
Contains general controls and pre-market approval
These devices cause high risks of danger to the patients.
For example; an oxygen support machine
What Is A Premarket Approval In Relation To FDA Regulations?
Premarket approval normally occurs when the manufacturer clears with the FDA.
This normally occurs when the manufacturer presents a new product.
The FDA will approve the product to penetrate the market and later will review the consumer feedback.
This allows them to determine if the product will pose danger to the consumers, before giving marketing approval.
When Is A Premarket Approval Required When Shipping FDA Regulated Products?
You will require a premarket approval while shipping products considered to pose a great danger when consumer user them.
For instance, if the owner is shipping medical devices that fall under class 3. They will require a premarket approval because they pose a great danger to patients.
What Is The Difference Between Premarket Approval And 510K Premarket Notification?
Both premarket approval and 510k premarket notification have distinguishing functions thus making them different.
Below are some of the main differences:
Premarket approval applies for totally new products in the market such as the class 3 medical devices.
Timeframe for premarket approval is strictly 6 months.
With premarket approval, the food and drug administration conduct thorough research to determining ease penetration of product to market.
This normally occurs the product poses great danger.
With remarket approval, the food and drug administration should approve or decline the product.
Premarket approval entails a lot of paper documentation that provides the plan for the establishment of these products.
On the other hand,
510k premarket notification applies when the manufacturer produces a similar product to that existing in the market.
The timeframe of submitting the 510k premarket notification is an average of 90 days which makes it faster and affordable.
Allows illustrations of the products to prove distinct features from the existing one
Also, 510k premarket notification does not require showcasing of all information.
There is an exemption if the manufacturer alters the products to perform the same functionalities.
Submitting 510k only guarantees the clearance process and therefore you cannot advertise it as food and drug administration approval.
How Long Does FDA Approval Take?

FDA Approved
Food and Drug Administration requires a certain timeframe for the complete approval process. This timeframe depends on the following;
Submission of the 510k applications
Submission of premarket approval
If you register individually.
Considering these factors FDA approval will take between 5 working days to 120 (equivalent to 8 months).
What Is The Difference Between ‘’FDA Registered’’ And ‘’FDA Approved’’ And ‘’FDA Cleared’’?
There are unique features that make each of them different features apply while evaluating what regulations govern them.
These differences include;
FDA approved implies a complete examination and review of products to ensure they are safe and effective for human consumption.
This means the products should not pose any health risks to the consumers.
FDA cleared applies where the manufacturer presents to the FDA a product that is like that existing in the market.
The FDA request for demonstrations to prove the product achieves the FDA regulations i.e., safety and effectiveness.
After a successful demonstration, the FDA will clear this product for marketing. This process is what we term as FDA cleared.
FDA requires the manufacturers to register with them before releasing their products to the markets.
Registration of the products allows the FDA to be aware of all activities you undertake to produce the products.
The main importance of registration is to help deal with any arising issues from malfunctioning devices.
What Is An FDA Hold In Shipping?
We can refer to food and drug administration as a form of custom hold at the custom clearance offices.
FDA hold occurs mainly when the FDA finds the need to hold products for further detailed examination.
The FDA will request more documentation to prove the usage of the products.
This decision is a result of goods not meeting the FDA threshold.
Therefore, the FDA will hold these goods for as long as the details are unclear.
Why Would The FDA Hold A Shipment?

Container yard
The key importance of the FDA is to evaluate and examine products for the safety of the consumers.
If this product does not meet these standards, FDA will hold these goods and send a holding notification to the relevant individuals.
The FDA holds this shipment if the owner refuses to provide proof that the product complies with the law.
Failure to provide a plan to bring the product into compliance
Failure to accept that the products violate the law.
How Long Can The FDA Hold A Package?
On normal occasions, FDA will hold this package for a period of 1 to 2 days, but under special circumstances, it can take even months.
The earlier the owner provides the information that the FDA request the faster they will release these packages.
Delays as a result owner withholding information will lead to a long period of holding.
What Happens When The FDA Holds A Shipment?
Normally, FDA holds products if they suspect that they need further inspection or provision of unclear information.
In case the FDA holds they will send a notification to the customs broker who has an obligation of notifying the owner.
The FDA will request additional information about the product
They might as well pick samples for lab testing
All these procedures help to determine if the products meet the US laws and regulations.
How Do I Perform An FDA Hold Status Check?
This status check is achievable by;
You can use the custom broker to follow up on the holding process
You can also contact the FDA via their platform.
This will require you to fill in your details in a form.
This form includes entry number, CPB, and FDA number.
THE FDA will communicate back to you.
What Can I Do If My Package Is Held By The FDA?
Normally if the food and drug administration hold the package, you should consider the following two options:
- Submit a request to allow you to make changes to the products to clear the violation issues.
- You can submit proof of the product’s information to the FDA to clear the violation. Submission of proof further allows the FDA to conduct a further examination.
What Is The FDA Product Code?
Food and Drug Administration product code provides a description of the products and what group they belong to.
FDA product code usually contains seven alphanumeric characters. It is made up of different elements such as;
- Product industry code
- Class code
- Subclass code
- Process indicator code
- Group code
What Is The Process For Importing FDA Regulated Products?
Importing products that under FDA regulations requires:
Involve custom brokers who act as third parties.
The ensure submission of relevant documents and make payments for all the entries.
The Customs broker will make an entry with Customs and Board Protection and later include an HTS.
The HTS consists of tags that assist food and drug administration which information is important and which is not.
The HTTUS code determines if the product meets the FDA standards. If it’s the case submission of all documents takes place.
Can A Customs Broker Help With FDA Custom Clearance?
Custom brokers act as agents between the buyer and the seller.
Thus, preventing direct involvement.
Customs brokers play a vital role in the customs clearance process.
Custom brokers help in the following ways;
They ensure that the goods you are importing adhere to FDA regulations
They determine a suitable harmonized tariff schedule which is a requirement by the customs and border protection.
Custom brokers provide you with FDA information and affirmation of compliance requirements.
During the entry of goods, they will always acquire a bond for you which help in the payment of the products.
They prepare and provide all the documentation to the food and drug administration.
They use Automated Broker Interface to transmit all information to FDA and back to the owner.
A customs broker can file a notice before.
They also provide answers to queries regarding FDA custom clearance.
What Is The Cost Of Importing FDA Regulated Products?
It is important to evaluate all possible cost options while importing FDA regulated products. Normally, these costs vary differently depending on the following factors:
The type of product you are importing into the country.
This means every product type you import will have costs differently normally due to sizes.
How much are the products worth?
This implies if the products constitute a high amount of money.
The cost charges will increase.
We can also determine the cost by the longevity period that the owner has been in business.
This means if the owner has been in business for long, they will pay less.
For start-up businesses, the costs are high which poses a challenge to the business owner.
Costs will vary from bond charges, shipping charges, and duties.
And it’s the responsibility of the business owner starting up to be aware and prepare for these costs.
In case you have problems with product quality inspection, BanSar is here to help.
Contact us now when importing from China.
The table below lists all official FDA Guidance Documents and other regulatory guidance. You can search for documents using key words, and you can narrow or filter your results by product, date issued, FDA organizational unit, type of document, subject, draft or final status, and comment period.
This feature is provided to give a convenient way to search for all FDA guidance documents from a single location.
If you cannot find the document you’re looking for here, you can browse separate collections of guidance documents by topic.
Go to Guidance Document Search
About FDA Guidance Documents
Guidance documents represent FDA’s current thinking on a topic. They do not create or confer any rights for or on any person and do not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations.
If you believe an FDA employee is not following FDA’s Good Guidance Practice regulations (21 CFR 10.115), you should contact the employee’s supervisor in the issuing office or Center. If the issue is not resolved, contact the next highest supervisor or the Center’s Ombudsman. If the issue is still not resolved, contact the FDA’s Office of the Ombudsman at:
FDA Office of the Ombudsman
10903 New Hampshire Avenue
WO Bldg. 1, room 4208
Silver Spring, MD 20993
Phone : 301-796-8530
Email: Ombuds@oc.fda.gov
Some Web links (URLs) embedded within guidance documents may have changed since the document was published. If you find a link that does not work, please try searching for the document using the document title. For more assistance, go to Contact FDA.
Commenting on Guidance Documents
Some FDA guidance documents on this list are indicated as open for comment. Although you can comment on any guidance at any time (see 21 CFR 10.115(g)(5)), to ensure that the Agency considers your comment on a draft guidance that is open for comments before it begins work on the final version of the guidance, submit either electronic or written comments by the closing date. Comments are submitted electronically through regulations.gov. For more information, see:
- How to Use Regulations.gov
- Find and Comment on FDA Dockets
Go to Class II Special Controls Documents
Guidance Document Search
Search
Some table information
Filters
Comment Closing Date on Draft*
| Summary | Document | Issue Date | FDA Organization | regulated product taxonomy hidden | Topic old hidden | Topic | Guidance Status | Open for Comment | Comment Closing Date on Draft | Docket Number | Guidance Type hidden | center taxonomy hidden | regulated product parent taxonomy hidden |
|---|
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов — Food and Drug Administration
Агентство Министерства здравоохранения и социальных служб США
| Обзор агентства | |
|---|---|
| Образован | 30 июня 1906 г.; 114 лет назад (1906-06-30) |
| Предыдущие агентства |
|
| Юрисдикция | Федеральное правительство США |
| Штаб-квартира | Кампус Уайт-Оук. 10903 Нью-Гэмпшир-авеню. Сильвер-Спринг, Мэриленд 20993. 39 ° 02′07 ″ с.ш., 76 ° 58′59 ″ з.д. / 39,03528 ° с.ш., 76,98306 ° Вт / 39,03528 ; -76.98306 Координаты : 39 ° 02′07 ″ N 76 ° 58′59 ″ з.д. / 39,03528 ° N 76,98306 ° W / 39,03528; -76,98306 |
| Сотрудники | 14 824 (2010) |
| Годовой бюджет | 3,16 миллиарда долларов (2020) |
| Руководители агентства |
|
| Материнское агентство | Департамент здравоохранения и социальных служб |
| Детские агентства |
|
| Веб-сайт | www.fda.gov |
The США Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA или USFDA ) является федеральным агентством при Департамента здравоохранения и социальных служб. FDA несет ответственность за защиту и укрепление общественного здоровья посредством контроля и надзора за безопасностью пищевых продуктов, табачными изделиями, пищевыми добавками, рецептурные и без рецепта фармацевтические препараты (лекарства), вакцины, биофармацевтические препараты, кровь переливание, медицинские устройства, устройства, излучающие электромагнитное излучение (ERED), косметика, корма и корма для животных и ветеринарные продукты.
Основное внимание FDA уделяет обеспечению соблюдения Федерального закона о пищевых продуктах, лекарствах и косметических средствах (FDC), но агентство также обеспечивает соблюдение других законов, в частности, Раздел 361 Закона о государственной службе здравоохранения, поскольку а также соответствующие правила. Большая часть этой работы по обеспечению соблюдения нормативных требований не связана напрямую с продуктами питания или лекарствами, но включает такие вещи, как регулирование лазеров, сотовых телефонов и презервативов, а также для борьбы с болезнями в различных контекстах: от домашних домашних животных до донорской спермы человека для использования в вспомогательной репродукции.
FDA возглавляет Уполномоченный по продовольствию и Препараты, назначенные Президентом с совета и согласия Сената . Комиссар подчиняется министру здравоохранения и социальных служб. Стивен М. Хан, доктор медицины, является нынешним комиссаром, по состоянию на декабрь 2019 года.
У FDA есть штаб-квартира в неинкорпорированном Уайт-Оук, Мэриленд. У агентства также есть 223 полевых офиса и 13 лабораторий, расположенных в 50 штатах, Виргинских островах США и Пуэрто-Рико. В 2008 году FDA начало направлять сотрудников в зарубежные страны, включая Китай, Индию, Коста-Рику, Чили, Бельгию и Соединенное Королевство.
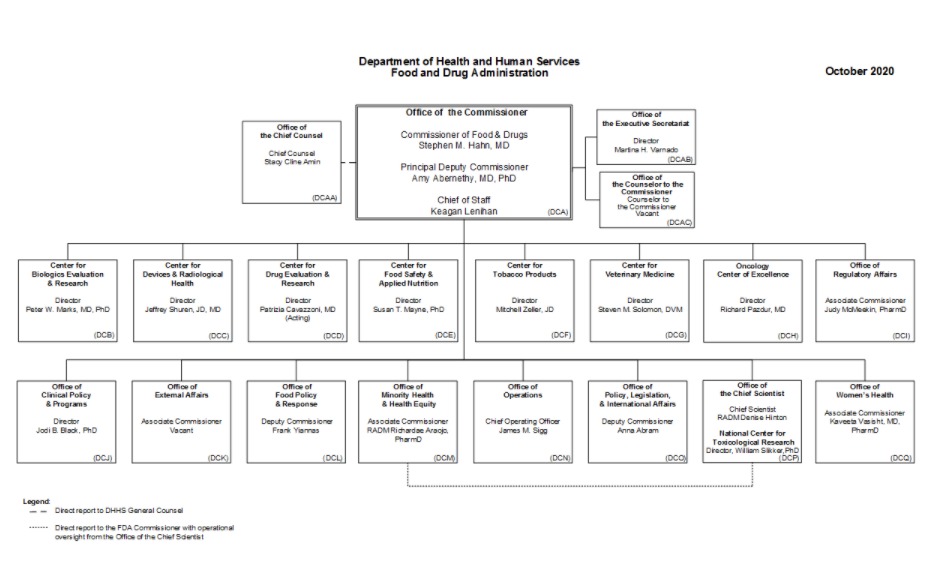
 В здании 31 FDA находится Офис Уполномоченного и Регулирующий орган Департамент здравоохранения и социальных служб. Агентство состоит из четырнадцати центров и офисов:
В здании 31 FDA находится Офис Уполномоченного и Регулирующий орган Департамент здравоохранения и социальных служб. Агентство состоит из четырнадцати центров и офисов:
Содержание
- 1 Организационная структура
- 2 Местоположение
- 2.1 Штаб-квартира
- 2.2 Федеральный исследовательский центр White Oak
- 2.3 Полевые местоположения
- 2.3.1 Управление по регуляторным вопросам
- 2.3.2 Управление по уголовным расследованиям
- 2.3.3 Другие места
- 3 Объем и финансирование
- 4 Регулирующие программы
- 4.1 Продукты питания и диетические добавки
- 4.1. 1 «Одобрено FDA» против «Принято FDA в пищевой промышленности»
- 4.2 Медицинские контрмеры (MCM)
- 4.3 Лекарства
- 4.3.1 Новые лекарства
- 4.3.1.1 Реклама и продвижение
- 4.3.1.2 Надзор за безопасностью после выхода на рынок
- 4.3.2 Дженерики
- 4.3.2.1 Скандал с непатентованными лекарствами
- 4.3.3 Безрецептурные препараты
- 4.3.4 Лечение лихорадки Эбола
- 4.3. 5 Тестирование на коронавирус (COVID-19)
- 4.3.1 Новые лекарства
- 4.4 Вакцины, кровь и тканевые продукты и биотехнологии
- 4.5 Медицинские и радиационно-излучающие устройства
- 4.5.1 «Одобрено FDA» против «Одобрено FDA»
- 4.6 Косметика
- 4.7 Ветеринарные товары
- 4.8 Тоба cco products
- 4.9 Регулирование живых организмов
- 4.10 Международное сотрудничество
- 4.1 Продукты питания и диетические добавки
- 5 Научные и исследовательские программы
- 6 Управление данными
- 7 История
- 7.1 Исторический первый: FDA и Opana ER Endo Pharmaceutical (2017)
- 8 Реформы 21 века
- 8.1 Инициатива Critical Path
- 8.2 Права пациентов на доступ к неутвержденным лекарствам
- 8.3 Постмаркетинговый мониторинг безопасности лекарственных средств
- 8.4 Тестирование педиатрических препаратов
- 8.5 Ваучер на приоритетное рассмотрение (PRV)
- 8.6 Правила для генерических биопрепаратов
- 8.7 Мобильные медицинские приложения
- 9 Критика
- 10 См. Также
- 11 Примечания
- 12 Ссылки
- 13 Дополнительная литература
- 14 Внешние ссылки ссылки
Организационная структура
 Таха Касс-Хаут (2014)
Таха Касс-Хаут (2014)
- Департамент здравоохранения и социальных служб
- Управление по санитарному надзору за качеством пищевых продуктов и медикаментов
- Управление комиссара
- Оперативное управление
- Управление равных возможностей трудоустройства
- Управление людских ресурсов
- Управление финансов, бюджета и снабжения
- Управление управления информацией и Технологии
- Управление информатики и технологических инноваций
- Директор: Таха А. Касс-Хаут (также занимает должность начальника Информатики здравоохранения FDA)
- Офис управления информацией
- Управление информатики и технологических инноваций
- Управление операций по обеспечению безопасности
- Управление инженерных сооружений и вспомогательных служб миссии
- Центр оценки и исследований биологических препаратов (CBER)
- Центр устройств и радиологического здоровья (CDRH)
- Центр оценки и исследований лекарственных средств (CDER)
- (OCE)
- Центр табачных изделий (CTP)
- Управление пищевых продуктов и ветеринарная медицина
- Центр ветеринарной медицины (CVM)
- Центр безопасности пищевых продуктов и прикладного питания (CFSAN)
- Управление глобальных регуляторных операций и политики (GO)
- Национальный центр токсикологических исследований (NCTR)
- Управление по нормативным вопросам
- Управление по санитарному надзору за качеством пищевых продуктов и медикаментов
Местоположение
 В здании 66 FDA находится Центр устройств и радиологического здоровья.
В здании 66 FDA находится Центр устройств и радиологического здоровья.
Штаб-квартира
Помещения штаб-квартиры FDA в настоящее время расположен в округе Монтгомери и округе Принс-Джордж в Мэриленде.
Федеральный исследовательский центр Уайт-Оук
С 1990 года у FDA есть сотрудники и на территории 130 акров (53 гектара) в районе Уайт-Оук в Силвер-Спринг, Мэриленд. В 2001 году Управление общих служб (GSA) начало новое строительство в кампусе, чтобы объединить 25 существующих подразделений FDA в столичном районе Вашингтона, его штаб-квартира в Роквилле, и несколько раздробленных офисных зданий. Первое здание, Лаборатория наук о жизни, было посвящено и открыто 104 сотрудниками в декабре 2003 года. По состоянию на декабрь 2018 года в кампусе FDA насчитывалось 10 987 сотрудников, размещенных на площади примерно 3 800 000 квадратных футов (350 000 квадратных метров), разделенных на десять офисных и четыре лабораторных корпуса. В кампусе находятся Офис Уполномоченного (OC), Управление по регуляторным вопросам (ORA), Центр оценки и исследований лекарственных средств (CDER), Центр Устройства и радиологическое здоровье (CDRH), Центр оценки и исследований биологических препаратов (CBER) и офисы Центра ветеринарной медицины (CVM).
С принятием закона FDA прогнозирует увеличение числа сотрудников на 64% до 18 000 в течение следующих 15 лет и хотело бы добавить приблизительно 1 600 000 квадратных футов (150 000 квадратных метров) офисных и специальных помещений к существующим помещениям. Национальная комиссия по капитальному планированию утвердила новый генеральный план этого расширения в декабре 2018 года, и ожидается, что строительство будет завершено к 2035 году, в зависимости от ассигнований GSA.
Расположение мест
 Лаборатория Арканзаса в Джефферсоне, Арканзас является штаб-квартирой Национального центра токсикологических исследований
Лаборатория Арканзаса в Джефферсоне, Арканзас является штаб-квартирой Национального центра токсикологических исследований
Управление по нормативным вопросам
Управление по вопросам регулирования считается «глазами и ушами» агентства, «проведение подавляющего большинства работ FDA в этой области. Его сотрудники, известные как сотрудники по безопасности потребителей, или, как более часто называют, следователи, инспектируют производственные и складские помещения, расследуют жалобы, заболевания или вспышки заболеваний и проверяют документацию в отношении медицинских устройств, лекарств, биологических продуктов и других предметов, если они может быть сложно провести физический осмотр или взять физический образец продукта. Управление по регуляторным вопросам разделено на пять регионов, которые делятся на 20 районов. Округа основаны примерно на географических подразделениях Федеральной судебной системы. Каждый округ состоит из главного районного офиса и ряда постов резидентов, которые представляют собой удаленные офисы FDA, обслуживающие конкретную географическую область. ORA также включает сеть регулирующих лабораторий Агентства, которые анализируют любые взятые физические пробы. Хотя образцы обычно связаны с пищевыми продуктами, некоторые лаборатории оснащены оборудованием для анализа лекарств, косметики и устройств, излучающих радиацию.
Управление уголовных расследований
 Ямайка, Квинс, региональное отделение штата Нью-Йорк — USFDA
Ямайка, Квинс, региональное отделение штата Нью-Йорк — USFDA
Управление уголовных расследований было создано в 1991 году для расследования уголовных дел. Для этого OCI нанимает около 200 специальных агентов по всей стране, которые, в отличие от следователей ORA, вооружены и не сосредотачиваются на технических аспектах регулируемых отраслей. Агенты OCI расследуют и раскрывают дела, в которых отдельные лица и компании совершали преступные действия, такие как мошеннические иски или сознательная и умышленная отправка известных фальсифицированных товаров в межгосударственной торговле. Во многих случаях OCI рассматривает дела, связанные с нарушениями Раздела 18 Кодекса Соединенных Штатов (например, сговор, ложные заявления, мошенничество с использованием электронных средств связи, мошенничество с использованием почты), в дополнение к запрещенным действиям, как определено в главе III Закона. Закон о FDC. Специальные агенты OCI часто происходят из других источников в сфере уголовных расследований и работают в тесном сотрудничестве с Федеральным бюро расследований, помощником генерального прокурора и даже Интерполом. OCI получает дела из различных источников, включая ORA, местные агентства и ФБР, и работает с ORA Investigators, чтобы помочь в разработке технических и научных аспектов дела.
Другие офисы
FDA имеет ряд других полевых офисов в Соединенных Штатах, помимо международных офисов в Китае, Индии, Европе, на Ближнем Востоке и в Латинской Америке.
Объем и финансирование
FDA регулирует потребительские товары на сумму более 2,4 триллиона долларов США, что составляет около 25% потребительских расходов в США. Это включает 466 миллиардов долларов на продажу продуктов питания, 275 миллиардов долларов на лекарства, 60 миллиардов долларов на косметику и 18 миллиардов долларов на витаминные добавки. Большая часть этих расходов приходится на товары, импортируемые в Соединенные Штаты; FDA отвечает за мониторинг импорта.
Запрос федерального бюджета FDA на 2012 финансовый год (FY) составил 4,36 миллиарда долларов, в то время как предлагаемый бюджет на 2014 год составляет 4,7 миллиарда долларов. Около 2 миллиардов долларов из этого бюджета формируются за счет сборов с пользователей. Фармацевтические фирмы оплачивают большую часть этих сборов, которые используются для ускорения рассмотрения лекарств. Запрос FDA в федеральный бюджет на 2008 финансовый год (с октября 2007 г. по сентябрь 2008 г.) составил 2,1 млрд долларов, что на 105,8 млн долларов больше, чем за 2007 финансовый год.
В феврале 2008 года FDA объявило, что Бюджетный запрос администрации Буша на 2009 финансовый год для агентства составлял чуть менее 2,4 миллиарда долларов: 1,77 миллиарда долларов на бюджетные полномочия (федеральное финансирование) и 628 миллионов долларов на оплату услуг. Запрошенные бюджетные полномочия были на 50,7 миллионов долларов больше, чем финансирование за 2008 финансовый год — примерно на три процента. В июне 2008 года Конгресс выделил агентству чрезвычайные ассигнования в размере 150 миллионов долларов на 2008 финансовый год и еще 150 миллионов долларов.
Программы регулирования
По состоянию на 2015 год агентство регулирует потребительские товары на сумму более 1 триллиона долларов., в том числе:
- 466 миллиардов долларов на продукты питания
- 275 миллиардов долларов на лекарства
- 60 миллиардов долларов на косметику
- 18 миллиардов долларов на витаминные добавки
Программы регулирования безопасности различаются широко в зависимости от типа продукта, его потенциальных рисков и регулирующих полномочий, предоставленных агентству. Например, FDA регулирует почти все аспекты отпускаемых по рецепту лекарств, включая тестирование, производство, маркировку, рекламу, маркетинг, эффективность и безопасность, однако регулирование FDA косметических средств в первую очередь сосредоточено на маркировке и безопасности. FDA регулирует большинство продуктов с помощью ряда опубликованных стандартов, соблюдение которых обеспечивается небольшим количеством проверок предприятий. Инспекционные наблюдения задокументированы в форме 483.
. В июне 2018 года FDA выпустило заявление о новых рекомендациях, которые помогут производителям продуктов питания и лекарств «реализовать защиту от потенциальных атак на поставки продовольствия в США». Одно из новых руководящих принципов включает правило о преднамеренном искажении (IA), которое требует от пищевой промышленности стратегий и процедур для снижения риска компрометации объектов и процессов, которые значительно уязвимы.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США также использует тактику позора со стороны регулирующих органов, в основном посредством публикации в Интернете сообщений о несоблюдении, предупреждающих писем и «списков позора». Регулирование посредством позора обуздывает чувствительность компаний к репутационному ущербу. Например, в 2018 году агентство опубликовало онлайн-«черный список», в котором назвало десятки фармацевтических компаний, которые якобы используют незаконные или неэтичные методы, чтобы помешать конкуренции со стороны компаний-производителей дженериков.
FDA часто работает с другими федеральными агентствами, включая Министерство сельского хозяйства, Управление по борьбе с наркотиками, Таможенно-пограничная служба и Безопасность потребительских товаров. Комиссия. Они также часто работают с местными правительственными учреждениями и органами власти штата при проведении проверок надзорных органов и принудительных мер.
Пищевые продукты и пищевые добавки
Регулирование пищевых продуктов и пищевых добавок Управлением по контролю за продуктами и лекарствами регулируется различными законами, принятыми Конгрессом США и интерпретируемыми FDA. В соответствии с Федеральным законом о пищевых продуктах, лекарствах и косметических средствах и сопутствующим законодательством FDA имеет право контролировать качество веществ, продаваемых в качестве пищевых продуктов в Соединенных Штатах, и отслеживать утверждения, сделанные на этикетке . как о составе, так и о пользе пищевых продуктов для здоровья.
FDA подразделяет вещества, которые оно регулирует как пищевые продукты, на различные категории, включая пищевые продукты, пищевые добавки, добавленные вещества (искусственные вещества, которые не вводятся в пищу намеренно, но, тем не менее, в нем), и БАДы. Пищевые добавки или диетические ингредиенты включают витамины, минералы, травы, аминокислоты и. ферменты. Конкретные стандарты упражнений FDA различаются от одной категории к другой. Кроме того, законодательство предоставило FDA ряд средств для устранения нарушений стандартов для данной категории веществ.
В соответствии с Законом о здоровье и образовании диетических добавок 1994 (DSHEA), FDA отвечает за обеспечение того, чтобы производители и дистрибьюторы пищевых добавок и диетических ингредиентов соответствовали текущим требованиям. Этим производителям и дистрибьюторам не разрешается рекламировать свои продукты фальсифицированным образом, и они несут ответственность за оценку безопасности и маркировку своей продукции.
У FDA есть «Консультативный список ингредиентов диетических добавок», который включает ингредиенты которые иногда появляются на пищевых добавках, но требуют дальнейшего изучения. Ингредиент добавляется в этот список, когда он исключен из использования в диетической добавке, не является одобренной пищевой добавкой или признан безопасным и / или подвергается требованию предварительного уведомления о продаже без удовлетворения требований..
«Одобрено FDA» vs. «Принято FDA в пищевой промышленности»
FDA не одобряет нанесение покрытий, используемых в пищевой промышленности. Не существует процесса проверки для утверждения состава антипригарных покрытий, а также FDA не проверяет и не тестирует эти материалы. Тем не менее, посредством управления процессами FDA имеет набор правил, регулирующих состав, производство и использование антипригарных покрытий. Следовательно, такие материалы, как политетрафторэтилен (тефлон) не являются и не могут рассматриваться как одобренные FDA, скорее, они «соответствуют требованиям FDA» или «приемлемы FDA».
Медицинские контрмеры (MCM)
Медицинские контрмеры (MCM) — это такие продукты, как биопрепараты и фармацевтические препараты, которые могут защищать или лечить последствия химической, биологической, радиологической или ядерной (CBRN) атаки. MCM также можно использовать для предотвращения и диагностики симптомов, связанных с атаками или угрозами CBRN. FDA реализует программу под названием «Инициатива по медицинскому противодействию FDA» (MCMi), которая финансируется федеральным правительством. Он помогает поддерживать «партнерские» агентства иорганизации в подготовке к чрезвычайным ситуациям в области здравоохранения, которые могут потребовать MCM.
Лекарства
 В здании 51 Управления по контролю за продуктами и лекарствами находится Центр оценки и исследований лекарственных средств.
В здании 51 Управления по контролю за продуктами и лекарствами находится Центр оценки и исследований лекарственных средств.
Центр оценки и исследований лекарственных средств использует разные требования к трем основные типам лекарственных средств: новыми лекарствами, непатентованным лекарствам, отпускаемым без рецепта. Лекарство считается «новым», если оно произведено другими средствами, использует вспомогательные вещества или неактивные ингредиенты, используемые для других целей или претерпевает какие-либо существенные изменения. Самые строгие требования предъявляются к новым молекулярным объектам: лекарствам, не основанным на лекарствах.
Новые лекарства
Новые лекарства перед утверждением FDA проходит тщательную проверку в рамках процесса, называемого заявкой на новое лекарство (NDA). При администрации Трампа агентство работало над ускорением утверждения лекарств. Критики, однако утверждают, что стандарты FDA недостаточно строги, что позволяет одобрять небезопасные или неэффективные лекарства. По умолчанию новые препараты отпускаются только по рецепту. Переход на безрецептурный (OTC) статус — это отдельный процесс, и сначала лекарство должно быть одобрено через NDA. Утвержденный стандарт считается «безопасным и при правильном применении».
Очень редко, ограниченные исключения из этого многоэтапного процесса, включающего испытания на животных и контролируемые клинические испытания. Так было во время эпидемии лихорадки Эбола 2015 года с применением рецепту и с разрешением ZMapp и других экспериментальных методов лечения, а также для новых препаратов, которые можно использовать для лечения изнурительных и / или очень редких состояний, при которых Ни одно из средств лечения или лекарств не является удовлетворительным, или в течение длительного периода времени не было никаких улучшений. Исследования становятся все длиннее, участвуют фармацевтические компании. Однако любые исключения из вышеупомянутого критерия подлежат строгому рассмотрению.
Реклама и продвижение
Управление FDA по продвижению рецептурных лекарств рассматривает и регулирует рекламу и продвижение рецептурных препаратов посредством надзора и выдачи исполнительных писем производителя фармацевтической продукции. Реклама и продвижение безрецептурных препаратов регулируется Федеральной торговой комиссией. FDA также предоставляет сторонним фирмам-исполнителям некоторый регуляторный надзор, например FDA ожидает, что сторонние поставщики и лаборатории соблюдают инструкции по охране здоровья и безопасности.
Положение о рекламе лекарств содержит два общих требования: (1) компания может рекламировать или продвигать препарат только по конкретным показаниям или медицинскому применению, для которого он был одобрен FDA. Кроме того, реклама должна содержать «справедливый баланс» между преимуществами и рисками (побочными эффектами) лекарства.
Термин не по назначению относится к использованию лекарств по показаниям, отличным от утвержденных FDA.
Надзор за безопасностью после выхода на рынок
После утверждения NDA спонсор должен проверять и сообщать в FDA о каждом возможном опыте пациента с лекарствами, о котором он узнает. Они должны сообщать о неожиданных серьезных и фатальных побочных эффектах в течение 15 дней, а о других событиях — ежеквартально. FDA также получает отчеты о побочных эффектах лекарств через свою программу MedWatch. Эти отчеты называются «спонтанными отчетами», потому что отчеты потребителей и медицинских работников являются добровольными.
Хотя это остается основным инструментом послепродажного надзора за безопасностью, требования FDA к постмаркетинговому управлению рисками растут. В качестве условий утверждения от спонсора может потребоваться проведение дополнительных клинических испытаний, называемых исследованийми фазы IV. В некоторых случаях FDA требует наличия планов управления рисками под названием Стратегии оценки и снижения рисков (REMS) для некоторых лекарств, требующих принятия мер для безопасного использования лекарственного средства. Например, талидомид может вызвать врожденные дефекты, но его применение перевешивает, если мужчины и женщины, принимающие наркотики, не зачать ребенка; программа REMS для талидомида требует проверяемого процесса, чтобы люди, принимающие препарат, предпримут действия, чтобы избежать беременности; Многие опиоидные препараты имеют программы REMS, чтобы избежать зависимости и утечки наркотиков. Препарат изотретиноин имеет программу REMS под названием iPLEDGE.
Дженерики
Дженерики являются химическими и терапевтическими эквивалентами фирменных препаратов, которые имеют истекший. Утвержденные дженерики должны иметь одинаковую дозировку, безопасность, эффективность, стабильность и качество, а также способ введения. В целом менее, чем их аналоги под маркой, производятся и продаются другие дороги, и в 1990-х годах на их долю приходилось около трети всех рецептов, выписанных в США. Для утверждения дженерика FDA требует научных доказательств того, что дженерик является взаимозаменяемым или терапевтически эквивалентным одобренному лекарству. Это называется Сокращенная заявка на новое лекарство (ANDA). По состоянию на 2012 год 80% всех одобренных FDA лекарств доступны в виде генериков.
Скандал с дженериками
В 1989 году разразился крупный скандал, связанный с процедурами, используемыми FDA для одобрения дженериков. для продажи населению. Обвинения в коррупции прирении непатентованных лекарств впервые возникли в 1988 году в обширном исследовании, проводимого Конгрессом США в FDA. Подкомитет по надзору по энергетике и торговле США возник на основании жалобы, поданной против FDA со стороны Mylan Laboratories Inc. из Питтсбурга. Когда его заявка на производство дженериков неоднократно откладывалась FDA, Mylan, частный расследование в отношении агентств в 1987 году. Mylan в итоге подал иск против бывших сотрудников FDA и четырех лекарств. производственные компании, обвиняемые в том, что коррупция в федеральном агентстве привела к рэкету и нарушению антимонопольного законодательства. «Порядок утверждения новых непатентованных лекарств был установлен сотрудниками FDA еще до того, как производители лекарств подали заявки», и, по словам Милана, эта незаконная процедура использовалась для предоставления льготного режима определенным компаниям. Летом 1989 г. трое экспертных лиц FDA (Чарльз И. Чанг, Дэвид Дж. Бранкато, Уолтер Клетч) признало себя виновными по уголовным обвинениям в получении взяток от производителей непатентованных лекарств, а также две компании (Par Pharmaceutical и ее дочерняя компания Quad Фармацевтика) признала себя виновной в даче взяток.
Кроме того, было обнаружено несколько производителей, представленных в поисках разрешения FDA на продажу соответствующих препаратов. Компания Vitarine Pharmaceuticals of New York, которая добивалась одобрения генерирующей версии препарата Диазид, лекарства от высокого кровяного давления, представила диазид, а не его генерическую версию, для тестирования FDA. В апреле 1989 года FDA провело расследование у 11 производителей на предмет нарушений; а позже довел это число до 13. Десятки лекарств были в конечном итоге приостановлены или отозваны производителями. В начале 1990-х годов Комиссия по ценным бумагам и биржам США предъявила предъявленные обвинения в мошенничестве с ценными бумагами против компании Bolar Pharmaceutical Company, крупного производителя дженериков, базирующегося в Лонг-Айленде, штат Нью-Йорк.
Лекарства, отпускаемые без рецепта
— встречные (безрецептурные) препараты, такие как аспирин, — это препараты и комбинации, которые не требуют рецепта врача. У FDA есть список из примерно 800 одобренных ингредиентов, которые комбинируют различные способы создания более 100 000 безрецептурных лекарственных препаратов. Многие ингредиенты, ранее были одобрены рецептурными препаратами, которые теперь считаются безопасными для использования без наблюдения практикующего врача, например, ибупрофен.
лечение Эболы
В 2014 году FDA добавило препарат Эбола, разработанный канадской фармацевтической компанией Tekmira, в программе Fast Track, но остановил испытания фазы 1 в июле в ожидании получения дополнительной информации о как действует препарат. Это стало особенно важным в условиях началась в конце марта 2014 г. в Западной Африке, которая началась в конце марта 2014 г. и закончилась в июне 2016 г.
Тестирование на коронавирус (COVID-19)
Во время пандемии коронавируса FDA предоставило разрешение на экстренное использование для средств индивидуальной защиты (СИЗ), диагностического оборудования in vitro, аппараты искусственной вентиляции легких и другие медицинские устройства.
18 марта инспекторы FDA отложили большинство инспекций иностранных предприятий и все внутренние инспекции учреждений эпиднадзора. В отличие от этого, Служба безопасности пищевых продуктов и инспекции (FSIS) USDA продолжила проверку мясокомбинатов, в результате чего 145 полевых сотрудников FSIS дали положительный результат на COVID-19 и трое умерли..
Вакцины, кровь и тканевые продукты, а также биотехнология
 Ученый FDA готовит образцы донорской крови для тестирования
Ученый FDA готовит образцы донорской крови для тестирования
Центр оценки и исследований биологических препаратов является ответвлением FDA. для обеспечения безопасности и эффективности биологических терапевтических агентов. К ним кровь кровь и продукты крови, вакцины, аллергены, продукты на клеточной и тканевой основе, а также продукты генной терапии. Новые биопрепараты должны проходить предмаркетинговый процесс утверждения, называемый заявкой на получение лицензии на биологические препараты (BLA), аналогичный процессуре для лекарств.
Первоначальные полномочия по государственному регулированию биологических продуктов были установлены Законом о контроле за биологическими препаратами 1902 года, дополнительные полномочия были установлены Законом о государственной службе здравоохранения 1944 года. Наряду с этими законами, Федеральный закон о пищевых продуктах, лекарствах и косметических продуктах распространяется также на все биологические продукты. Первоначально организация, ответственная за регулирование биологических продуктов, находилась в ведении Национальных институтов здравоохранения ; эти полномочия были переданы FDA в 1972 году.
Медицинские и радиационно-излучающие устройства
 В здании 62 FDA размещено Центр устройств и радиологического здоровья.
В здании 62 FDA размещено Центр устройств и радиологического здоровья.
Центр устройств и радиологического здоровья (CDRH) — это подразделение FDA, ответственное за предварительное одобрение всех медицинских устройств, а также надзор за производством, производственной и безопасностью этих устройств. Определение медицинского устройства дано в Законе FDC и включает простые зубные щетки до сложных устройств, таких как имплантируемые нейростимуляторы. CDRH также контролирует показатели безопасности немедицинских устройств, которые испускают тип электромагнитного излучения. Примеры устройств, регулируемых CDRH, включают сотовые телефоны, оборудование для досмотра багажа в аэропорту, телевизионные приемники, микроволновые печи, кабины для загара. и лазерные продукты..
Регулирующие полномочия CDRH включают в себя требования технических отчетов от производителей или импортеров регулируемых продуктов, чтобы излучающие продукты соответствовали обязательным стандартам безопасности, для декларирования регулируемых продуктов с дефектом, а также для отзыва дефектных продуктов. CDRH также проводит ограниченное количество прямых испытаний продукции.
«Одобрено FDA» и «Одобрено FDA»
Запросы на разрешение к медицинским устройствам, которые доказывают, что они «в степени эквивалентны» предикатным устройствам, уже имеющимся на рынке. «Утвержденные новые требования к безопасности и эффективности», например, они могут быть проверены на безопасность в случае использования токсичных опасностей. Оба элемента должны быть подтверждены.
Косметика
Косметика регулируется Центром безопасности пищевых продуктов и прикладного питания, тот же филиал FDA, который регулирует пищу. Косметические продукты, как правило, не подлежат предварительному одобрению FDA, за исключением тех случаев, когда они делают их лекарственные препараты (см. Космецевтика ). Тем не менее, все красящие добавки должны быть одобрены FDA, прежде чем производители Другие их в косметические продукты, продаваемые в США. FDA регулирует маркировку косметических средств, и косметика, не прошедшая испытания на безопасность, должна иметь предупреждение.
Согласно отраслевой группе защиты Американский совет по науке и здоровью, хотя косметическая промышленность несет основную ответственность за обеспечение безопасности своей продукции, FDA также имеет право вмешиваться, когда необходимо для защиты населения, но в целом не требует предварительного утверждения или тестирования. В ACSH говорится, что компании должны размещать предупреждающие надписи на своих продуктах, если они не были протестированы, и что эксперты по косметическим ингредиентам также играют роль в мониторинге безопасности, влияя на их использование, но также не имеют юридических полномочий. Согласно ACSH, в целом организация проверила около 1200 ингредиентов и предложила ограничить несколько сотен, но не существует стандартного или системного метода проверки химических веществ на предмет безопасности и четкого определения того, что подразумевается под « безопасностью », чтобы все химические вещества тестируются на той же основе.
Ветеринарные продукты
Центр ветеринарной медицины (CVM) является центром FDA, который регулирует пищевые добавки и лекарства, которые дан животным. CVM регулирует лекарства для животных, корм для животных, включая домашних животных, и медицинские устройства для животных. Требования FDA по предотвращению распространения губчатой энцефалопатии крупного рогатого скота также выполняются CVM через инспекции производителей кормов. CVM не регулирует вакцины для животных; ими занимается Министерство сельского хозяйства США.
Табачные изделия
FDA регулирует табачные изделия с полномочиями, установленными в 2009 г. Профилактика курения в семье и борьба против табака. Акт. Этот закон требует цветных предупреждений на пачках сигарет и печатной рекламы, а также текстовых предупреждений из США. Главный хирург.
FDA объявило о девяти новых графических предупредительных этикетках в июне 2011 года, и их планируется разместить на упаковке к сентябрю 2012 года. Дата внедрения неизвестна из-за продолжающихся разбирательств по делу Р.Дж. Reynolds Tobacco Co. против Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США. R.J. Рейнольдс, Lorillard, Commonwealth Brands, Liggett Group и Santa Fe Natural Tobacco Company подали иски в Вашингтон., Федеральный суд округа Колумбия, утверждая, что графические надписи являются неконституционным способом принуждения табачных компаний к участию в пропаганде борьбы с курением от имени правительства.
A Первая поправка юрист, Флойд Абрамс, представляет табачные компании в этом деле, утверждая, что требование наличия графических предупреждающих надписей на законном продукте не может выдержать конституционного контроля. Ассоциация национальных рекламодателей и Американская рекламная федерация также подали краткую информацию по иску, утверждая, что эти ярлыки нарушают коммерческую свободу слова и могут привести к дальнейшему вмешательству правительства, если их не оспаривать.. В ноябре 2011 года федеральный судья Ричард Леон Окружного суда США по округу Колумбия временно приостановил выпуск новых этикеток, вероятно, отсрочив требование о том, чтобы табачные компании отображали этикетки. США Верховный суд в конечном итоге мог решить этот вопрос.
В июле 2017 года FDA объявило план, который снизит текущие уровни никотина, разрешенного в табачных сигаретах.
Регулирование живых организмов
С принятием предварительного уведомления 510 (k) k033391 в январе 2004 года FDA предоставило доктору Рональду Шерману разрешение на производство и продажу медицинских личинок для использования на людях или других животных в качестве рецептурных медицинских препаратов. устройство. Медицинские личинки представляют собой первый живой организм, разрешенный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов для производства и продажи в качестве медицинского устройства, отпускаемого по рецепту.
В июне 2004 года FDA одобрило Hirudo medicinalis (медицинские пиявки) как второй живой организм, который будет использоваться в качестве медицинского устройства.
FDA также требует, чтобы молоко было пастеризовано для удаления бактерий.
Международное сотрудничество
В феврале 2011 года Президент Барак Обама и премьер-министр Канады Стивен Харпер выпустили «Декларацию об общем видении безопасности периметра и экономической конкурентоспособности» и объявили о создании Совета по сотрудничеству в области регулирования между Канадой и США (RCC) «для повышения прозрачности регулирования и низкой между двумя странами».
Согласно мандату RCC, FDA и Health Canada предпринял «первое в своем роде» инициативу, выбрав «в качестве первой области согласования показаний к простуде для некоторых безрецептурных антигистаминных ингредиентов (GC 2013-01-10)».
Более недавним примером международной работы FDA является их сотрудничество в 2018 году с регулирующими и правоохранительными органами по всему миру через Интерпол в рамках операции Pangea XI. FDA нацеле но на 465 веб-сайтов, которые незаконно продавали потенциально опасные, неутвержденные версии опиоидов, онкологических и противовирусных рецептурных препаратов потребителям из США. Агентство сосредоточилось на схемах отмывания транзакций, чтобы раскрыть сложную сеть наркотиков в Интернете.
Научные и исследовательские программы
 Лаборатория FDA в здании 64 в Силвер-Спринг, Мэриленд
Лаборатория FDA в здании 64 в Силвер-Спринг, Мэриленд
FDA проводит исследования и разработки для разработки технологий и стандартов, которые поддерживают его регулирующую роль, с целью решения научных и технических проблем, прежде чем они станут препятствиями. Исследования FDA включают в себя области биопрепаратов, медицинских устройств, лекарств, женского здоровья, токсикологии, безопасности пищевых продуктов и прикладного питания, а также ветеринарии.
Управление данными
FDA собрало большое количество объем данных за десятилетия. В марте 2013 года был создан OpenFDA, чтобы обеспечить легкий доступ к данным для общественности, и он был официально запущен в июне 2014 года.
История
Вплоть до 20-го века, было несколько федеральных законов, регулирующих содержание и продажу продуктов питания и фармацевтических препаратов отечественного производства, за одним исключением был недолговечный Закон о вакцинах 1813 года. История FDA восходит к концу 19 века и США. Министерство сельского хозяйства, позже его Бюро химии. При Харви Вашингтоне Вили, назначенном главным химиком в 1883 году, Отдел начал проводить исследования фальсификации и неправильного брендинга пищевых продуктов. и лекарства на американском рынке. Пропаганда Уайли возникла в то время, когда общественность была возбуждена опасностями на рынке из-за разоблачения журналистов, таких как Аптон Синклер, и стала частью общей тенденции к ужесточению федерального законодательства в соответствующих вопросах. общественной безопасности в Эру Прогресса. Закон о контроле за биологическими препаратами 1902 года был принят после того, как дифтерийный антитоксин, полученный из сыворотки, зараженной столбняком, был использован для производства вакцины, которая стала причиной смерти тринадцати детей в Санкт-Петербурге. Луис, штат Миссури. Первоначально сыворотка была получена от лошади по имени Джим, которая заразилась столбняком.
 Харви В. Уили, главный защитник Закона о пищевых продуктах и лекарствах
Харви В. Уили, главный защитник Закона о пищевых продуктах и лекарствах
В июне 1906 года президент Теодор Рузвельт подписал закон Закон о пищевых продуктах и лекарствах 1906 года, также известный как «Закон Уайли» по имени его главного защитника. Закон запрещал под угрозой конфискации товаров межгосударственную перевозку «фальсифицированных» продуктов питания. Закон применял аналогичные наказания к межгосударственному маркетингу «фальсифицированных» лекарств, в которых «стандарт силы, качества или чистоты» активного ингредиента не был четко указан на этикетке или перечислен в Фармакопее США. или Национальный формуляр.
Ответственность за проверку продуктов питания и лекарств на предмет «фальсификации» или «неправильного брендинга» была возложена на Химическое бюро Wiley USDA. Уайли использовал эти новые регулирующие полномочия для проведения агрессивной кампании против производителей пищевых продуктов с химическими добавками, но вскоре авторитет Химического бюро был проверен судебными решениями, которые узко определяли полномочия бюро и устанавливали высокие стандарты для доказательства мошенничества. В 1927 году регулирующие полномочия Химического бюро были реорганизованы в новый орган Министерства сельского хозяйства США — Управление по контролю за продуктами питания, лекарствами и инсектицидами. Три года спустя это название было сокращено до Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA).
К 1930-м годам разоблачающие журналисты, организации по защите потребителей и федеральные регулирующие органы начали проводить кампанию за усиление нормативных требований. опубликовав список вредных продуктов, которые были признаны допустимыми в соответствии с законом 1906 года, в том числе радиоактивные напитки, тушь приманка для ресниц, вызывающая слепоту, и бесполезные «лекарства» от диабета и туберкулеза. Предложенный закон не мог пройти через Конгресс Соединенных Штатов в течение пяти лет, но был быстро принят в качестве закона после общественного протеста по поводу трагедии эликсир сульфаниламида 1937 года, в которой более 100 человек умерли в результате применения препарата, созданного на основе токсичного, непроверенного растворителя.
Президент Франклин Делано Рузвельт подписал Федеральный закон о пищевых продуктах, лекарствах и косметических средствах в качестве закона 24 июня 1938 г. Новый закон значительно усилил федеральные регулирующие полномочия в отношении лекарств, обязывая проводить предпродажную проверку безопасности всех новых лекарств, а также запрещая ложные терапевтические заявления в маркировке лекарств, не требуя, чтобы FDA доказало мошеннический умысел. Вскоре после принятия Закона 1938 года FDA начало определять определенные лекарства как безопасные для использования только под наблюдением медицинского работника, а категория «только по рецепту » была надежно закреплена в законе Поправка Дарема-Хамфри в 1951 году. Эти разработки подтвердили широкие полномочия FDA по обеспечению постмаркетингового отзыва неэффективных лекарств.
 Медицинский сотрудник Александр Флеминг, доктор медицины, исследует часть 240-тома применение новых лекарств в конце 1980-х. После введения в действие поправки к лекарственным средствам 1962 г. количество применений значительно выросло.
Медицинский сотрудник Александр Флеминг, доктор медицины, исследует часть 240-тома применение новых лекарств в конце 1980-х. После введения в действие поправки к лекарственным средствам 1962 г. количество применений значительно выросло.
За пределами США продавался препарат талидомид для облегчения общей тошноты и утреннего недомогания, но вызывал врожденные дефекты и даже смерть тысяч младенцев при приеме во время беременности. Американских матерей это не коснулось, поскольку доктор Фрэнсис Олдхэм Келси из FDA отказалась разрешить продажу лекарства. В 1962 году была принята поправка Кефовера-Харриса к Закону о FDC, которая произвела «революцию» в регулирующем органе FDA. Самым важным изменением было требование, чтобы все заявки на новые лекарственные средства демонстрировали «существенные доказательства» эффективности лекарственного средства по показаниям на рынке в дополнение к существующему требованию о предпродажной демонстрации безопасности. Это ознаменовало начало процесса утверждения FDA в его современной форме.
Эти реформы привели к увеличению времени и трудностей, необходимых для вывода лекарства на рынок. Одним из наиболее важных законодательных актов в создании современного американского фармацевтического рынка был Закон 1984 о конкуренции цен на лекарства и восстановлении срока действия патентов, более известный как «Закон Хэтча-Ваксмана» по имени его главных спонсоров. Закон расширил срок действия патента на новые лекарства и частично увязал это продление с продолжительностью процесса утверждения FDA для каждого отдельного лекарства. Для производителей дженериков Закон создал новый механизм утверждения, Сокращенную заявку на новое лекарство (ANDA), в которой производителю дженериков нужно только продемонстрировать, что их генерический препарат имеет тот же самый активный ингредиент, путь введения, лекарственная форма, сила и фармакокинетические свойства («биоэквивалентность») в качестве соответствующего фирменного препарата. Этому Закону приписывают, по сути, создание современной индустрии непатентованных лекарств.
Обеспокоенность по поводу продолжительности процесса утверждения лекарств была выдвинута на первый план в начале эпидемии СПИДа. В середине и конце 1980-х годов ACT-UP и другие организации активистов ВИЧ обвинили FDA в необоснованной задержке утверждения лекарств для борьбы с ВИЧ и оппортунистическими инфекциями. Отчасти в ответ на эту критику FDA выпустило новые правила для ускорения утверждения лекарств от опасных для жизни заболеваний и расширило доступ к лекарствам до утверждения для пациентов с ограниченными вариантами лечения. Все первоначальные препараты, одобренные для лечения ВИЧ / СПИДа, были одобрены через эти ускоренные механизмы утверждения. Фрэнк Янг, в то время уполномоченный FDA, стоял за Фазой II Плана действий, разработанной в августе 1987 года для более быстрого одобрения лекарств от СПИДа.
В двух случаях правительства штатов пытались легализовать лекарства, которые не были утверждены FDA. утвержден. В соответствии с теорией, согласно которой федеральный закон, принятый в соответствии с конституционными полномочиями, отменяет противоречащие друг другу законы штатов, федеральные власти по-прежнему претендуют на право изымать, арестовывать и преследовать в судебном порядке за хранение и продажу этих веществ, даже в тех штатах, где они разрешены законодательством штата. Первой волной стала легализация лаэтрила в 27 штатах в конце 1970-х годов. Этот препарат использовался для лечения рака, но научные исследования как до, так и после этой законодательной тенденции показали, что он неэффективен. Вторая волна касалась медицинской марихуаны в 1990-е и 2000-е годы. Хотя Вирджиния приняла закон, разрешающий врачам рекомендовать каннабис при глаукоме или побочных эффектах химиотерапии, более широкая тенденция началась в Калифорнии с Закона о милосердном использовании от 1996 года.
Исторические первые: FDA и Endo Opana ER компании Pharmaceutical (2017)
Когда 8 июня 2017 года FDA обратилось к Endo Pharmaceuticals с просьбой удалить с рынка оксиморфон гидрохлорид, это был первый такой запрос. в истории FDA.
Реформы 21 века
Инициатива Critical Path
Инициатива Critical Path — это попытка FDA стимулировать и способствовать национальным усилиям по модернизации науки, с помощью которых FDA -регулируемые продукты разрабатываются, оцениваются и производятся. Инициатива была запущена в марте 2004 г., когда был выпущен отчет «Инновации / застой: проблемы и возможности на критическом пути к новым медицинским продуктам».
Права пациентов на доступ к неутвержденным лекарствам
Программа «Милосердные исследования новых наркотиков» была создана после того, как в 1978 году Рэндалл против США вынес решение в пользу Роберта С. Рэндалла, создав программу для медицинской марихуаны.
A 2006 судебное дело Эбигейл Альянс против фон Эшенбаха привело бы к радикальным изменениям в регулировании FDA в отношении неразрешенных препаратов. Альянс Абигейл утверждал, что FDA должно лицензировать лекарства для использования неизлечимо больными пациентами с «отчаянными диагнозами» после того, как они завершат тестирование Фазы I. Дело выиграло первоначальную апелляцию в мае 2006 года, но это решение было отменено повторным слушанием в марте 2007 года. Верховный суд США отказался рассматривать дело, а окончательное решение отрицало наличие права на использование неутвержденных лекарств.
Критики регулятивной власти FDA утверждают, что FDA слишком много времени занимает одобрение лекарств, которые могут облегчить боль и страдания людей быстрее, если они будут выпущены на рынок раньше. Кризис со СПИДом потребовал определенных политических усилий по упорядочению процесса утверждения. Однако эти ограниченные реформы были нацелены на лекарства от СПИДа, а не на более широкий рынок. Это привело к призыву к более надежным и устойчивым реформам, которые позволили бы пациентам под наблюдением их врачей получать доступ к лекарствам, прошедшим первый раунд клинических испытаний.
Постмаркетинговый мониторинг безопасности лекарств
Широко разрекламированный отзыв Виокса, нестероидного противовоспалительного препарата (НПВП), который, по оценкам, способствовал сердечным приступам со смертельным исходом у тысяч американцев сыграла важную роль в продвижении новой волны реформ безопасности как на уровне нормотворчества FDA, так и на законодательном уровне. Vioxx был одобрен FDA в 1999 году и первоначально предполагалось, что он будет более безопасным, чем предыдущие НПВП, из-за снижения риска кровотечения из кишечного тракта. Тем не менее, ряд до- и постмаркетинговых исследований показали, что Vioxx может увеличивать риск инфаркта миокарда, и это было окончательно продемонстрировано результатами исследования APPROVe в 2004 году.
Столкнувшись с многочисленными судебными исками, производитель добровольно снял его с рынка. Пример Vioxx стал ярким в продолжающихся дебатах о том, следует ли оценивать новые препараты на основе их абсолютной безопасности или их безопасности относительно существующих методов лечения данного состояния. После отзыва Vioxx в крупных газетах, медицинских журналах, организациях по защите прав потребителей, законодателях и должностных лицах FDA раздались многочисленные призывы к реформированию процедур FDA для регулирования безопасности лекарственных средств до и после продажи.
В 2006 г. комитет Конгресса был назначен Институтом медицины для обзора правил фармацевтической безопасности в США и выпуска рекомендаций по их улучшениям. Комитет состоял из 16 экспертов, в том числе лидеров в области клинической медицины, медицинских исследований, экономики, биостатистики, права, государственной политики, общественного здравоохранения и смежных медицинских профессий, а также нынешних и бывших руководителей из фармацевтика, больница и страхование здоровья. Авторы обнаружили серьезные недостатки в действующей системе FDA для обеспечения безопасности лекарств на американском рынке. В целом авторы призвали к увеличению регулирующих полномочий, финансирования и независимости FDA. Некоторые из рекомендаций комитета были включены в проекты поправки PDUFA IV, которая была подписана как Закон 2007 года о поправках к Управлению по контролю за продуктами и лекарствами..
С 2011 года были созданы планы действий по минимизации рисков (RiskMAPS). чтобы риски, связанные с лекарством, никогда не перевешивали преимущества этого лекарства в постмаркетинговый период. Эта программа требует от производителей разработки и проведения периодических оценок эффективности своих программ. Планы действий по минимизации рисков разрабатываются в зависимости от общего уровня риска, который рецептурный препарат может представлять для населения.
Тестирование на наркотики у детей
До 1990-х годов только 20% всех лекарств, назначаемых детям в Соединенных Штатах, были протестированы на безопасность и эффективность в педиатрической популяции. Это стало серьезной проблемой для педиатров, поскольку накопились доказательства того, что физиологическая реакция детей на многие лекарства значительно отличается от воздействия этих лекарств на взрослых. Дети по-разному реагируют на лекарства по многим причинам, включая размер, вес и т. Д. Было несколько причин, по которым было проведено мало медицинских испытаний с детьми. Для многих лекарств дети составляли настолько небольшую часть потенциального рынка, что производители лекарств не считали такое тестирование рентабельным.
Кроме того, считалось, что дети этически ограничены в своей способности давать информированное согласие, возникло больше препятствий со стороны государственных и институциональных органов для утверждения этих клинических испытаний, а также возросла озабоченность по поводу юридической ответственности. Таким образом, на протяжении десятилетий большинство лекарств, прописываемых детям в США, делалось не одобренным FDA, «не по назначению», с дозировками, «экстраполированными» из данных для взрослых посредством расчетов массы тела и площади поверхности тела.
Первой попыткой FDA решить эту проблему стало принятое FDA Окончательное правило 1994 года о педиатрической маркировке и экстраполяции, которое позволяло производителям добавлять информацию о педиатрической маркировке, но требовало лекарств, которые не были протестированы на педиатрическую безопасность и эффективность. нести отказ от ответственности по этому поводу. Однако это правило не побудило многие фармацевтические компании к проведению дополнительных исследований педиатрических препаратов. В 1997 году FDA предложило правило, требующее проведения испытаний педиатрических лекарств от спонсоров новых заявок на лекарства. Однако это новое правило было успешно отменено в федеральном суде как превышающее установленные законом полномочия FDA.
Пока разворачивались эти дебаты, Конгресс использовал Закон о модернизации Управления по санитарному надзору за качеством пищевых продуктов и медикаментов 1997 года для принятия стимулов. это дало производителям фармацевтической продукции продление срока действия патента на шесть месяцев на новые лекарства, представленные с данными педиатрических испытаний. Закон о лучших фармацевтических препаратах для детей от 2007 года повторно санкционировал эти положения и разрешил FDA запрашивать спонсируемое NIH тестирование для тестирования педиатрических лекарственных препаратов, хотя эти запросы подлежат ограничению финансирования NIH. В Конгрессе Конгресс закрепил полномочия FDA требовать проведения спонсируемых производителем испытаний педиатрических препаратов для определенных препаратов в качестве «крайней меры», если стимулы и механизмы государственного финансирования окажутся неадекватными.
Ваучер на приоритетное рассмотрение (PRV)
ваучер на приоритетное рассмотрение является положением Закона о внесении поправок в Управление по санитарному надзору за качеством пищевых продуктов и медикаментов от 2007 года, который предоставляет передаваемый «ваучер на приоритетное рассмотрение» любой компании, получившей одобрение на лечение. для забытых тропических болезней. Система была впервые предложена преподавателями Университета Дьюка Дэвидом Ридли, Генри Грабовски и Джеффри Мо в их статье Health Affairs 2006 года: «Разработка лекарственных препаратов для развивающихся стран». Президент Обама подписал закон Закон о безопасности и инновациях Управления по санитарному надзору за качеством пищевых продуктов и медикаментов 2012 года, который продлил действие разрешения до 2017 года.
Правила для генерических биопрепаратов
С 1990-х годов многие новыми успешными лекарствами для лечения рака, аутоиммунных заболеваний и других состояний были белковые препараты биотехнологии, регулируемые Центр оценки и исследований биологических препаратов. Многие из этих лекарств чрезвычайно дороги; например, противораковый препарат Авастин стоит 55000 долларов в год, а заместительная ферментная терапия препарат Церезим стоит 200000 долларов в год и должен быть принимается болезнью Гоше пациентами на всю жизнь.
Биотехнологические препараты не имеют простых, легко поддающихся проверке химических структур обычных лекарств и производятся с помощью сложных, часто патентованных, методов, таких как трансгенные культуры клеток млекопитающих. Из-за этих сложностей Закон Хэтча-Ваксмана 1984 года не включил биопрепараты в процесс Сокращенной заявки на новое лекарство (ANDA). Это исключило возможность конкуренции дженериков за биотехнологические препараты. В феврале 2007 года идентичные законопроекты были внесены в Палату представителей, чтобы создать процесс ANDA для утверждения генерических биопрепаратов, но не были приняты.
Мобильные медицинские приложения
В 2013 году было выпущено руководство для регулирования мобильных медицинских приложений и защиты пользователей от их непреднамеренного использования. В этом руководстве различаются приложения, подпадающие под действие нормативных требований, на основе маркетинговых заявлений приложений. Включение руководящих принципов на этапе разработки этих приложений было предложено для ускорения выхода на рынок и егоутверждения.
Критика
FDA осуществляет регулирующий надзор за большим количеством продуктов, которые влияют на здоровье и жизнь американских граждан. В результате полномочия и решения FDA тщательно контролируются несколькими правительственными и неправительственными организациями. В отчете Института медицины 2006 года о фармацевтическом регулировании в США за 1,8 миллиона долларов были обнаружены серьезные недостатки в существующей системе FDA для обеспечения безопасности лекарств на американском рынке. В целом авторы призвали к усилению регулирующих полномочий, финансирования и независимости FDA.
Девять ученых FDA обратились к тогдашнему избранному президенту Бараку Обаме из-за давления со стороны руководства, испытанного во время президентства Джорджа Буша, с целью манипулирования данными, в том числе в отношении процесса проверки медицинских устройств. Эти опасения, характеризуемые как «коррумпированные и искаженные нынешними менеджерами FDA, тем самым подвергающие риску американский народ», также были подчеркнуты в отчете агентства за 2006 год.
FDA также подвергалось критике с противоположной стороны. точка зрения, как слишком жесткая по отношению к промышленности. Согласно анализу, опубликованному на веб-сайте либертарианской Mercatus Center, многие считают, что FDA выходит за рамки своих регулирующих полномочий и подрывает малый бизнес и мелкие фермы в пользу крупных корпораций. Три из ограничений FDA в рамках их анализа — это разрешение на выпуск новых лекарств и устройств, контроль над произношением производителей и введение требований по рецептам. Авторы утверждают, что на все более сложном и разнообразном рынке пищевых продуктов FDA не имеет возможности адекватно регулировать или проверять пищевые продукты.
Однако есть показатель того, что FDA может быть слишком слабым в процессе утверждения, в частности для медицинских устройств, исследование 2011 года, проведенное доктором Дайаной Цукерман и Полом Брауном из Национального исследовательского центра для женщин и семей, и доктором Стивеном Ниссеном из Клиника Кливленда, опубликованная в Архиве внутренней медицины, показала, что большинство медицинских устройств, отозванных за последние пять лет из-за «серьезных проблем со здоровьем или смерти», были ранее одобрены FDA с использованием менее строгий и дешевый процесс 510 (k). В некоторых случаях устройства считались настолько низкоопасными, что не нуждались в регулировании FDA. Из 113 отозванных устройств 35 предназначались для сердечно-сосудистых заболеваний.
См. Также
- Побочная реакция
- Неблагоприятное событие
- Неблагоприятная реакция на лекарство
- Биозащита
- Биозащита в США
- Проведение исследования эффективности лекарств
- Закон о модернизации Управления по санитарному надзору за качеством пищевых продуктов и медикаментов 1997 г.
- Закон FDA о модернизации безопасности пищевых продуктов 2011 г.
- Программа развития FDA Fast Track (для лекарств)
- Закон 2007 года о поправках к Управлению по санитарному надзору за качеством пищевых продуктов и медикаментов (например, лекарства)
- Закон о безопасности и инновациях Управления по контролю за продуктами и лекарствами 2012 года (GAIN / QIDP и т. Д.)
- Закон об обратной выгоде
- Исключение для исследуемых устройств (для использования в клинических испытаниях)
- Поправка Кефовера-Харриса 1962 г. — требуется «доказательство эффективности» лекарств
Международный:
- Управление пищевых продуктов
- Международная конференция по гармонизации технических требований к регистрации лекарственных средств для использования человеком (ICH)
- Austr alia: Управление терапевтических товаров
- Бразилия: Национальное агентство по надзору за здоровьем
- Канада: Управление по продаже товаров медицинского назначения
- Канада: Министерство здравоохранения Канады
- Дания: Датское агентство по лекарственным средствам
- Европейский союз: Европейское агентство по лекарственным средствам
- Германия: Федеральный институт лекарственных средств и медицинских устройств
- Индия: Управление по безопасности и стандартам пищевых продуктов Индии
- Индия: Центральная организация по контролю за стандартами лекарственных средств
- Япония: Министерство здравоохранения, труда и социального обеспечения (MHLW)
- Япония: Агентство по фармацевтике и медицинскому оборудованию
- Мексика: Федеральная комиссия по защите от санитарных рисков
- Сингапур: Управление здравоохранения
- Соединенное Королевство: Агентство по регулированию лекарственных средств и товаров медицинского назначения
- США: Управление по санитарному надзору за качеством пищевых продуктов и медикаментов
Примечания
Ссылки
Дополнительная литература
- Гивель, Майкл (декабрь 2005 г.). «Гамбит FDA Филиппа Морриса: полезно для общественного здравоохранения?» Журнал политики общественного здравоохранения (26): стр. 450–468
- Хеннингер, Дэниел (2002). «Отставание от лекарств». В Дэвид Р. Хендерсон (ред.). Краткая энциклопедия экономики (1-е изд.). Библиотека экономики и свободы.OCLC 317650570, 50016270, 163149563
- Хилтс, Филип Дж. (2003). Защита здоровья Америки: FDA, бизнес и сто лет регулирования. Нью-Йорк: Альфред Э. Кнопф. ISBN 0-375-40466-X
- Кевин Файн, Мэтью Добресс, Г. Калеб Александр (2013). «Закон о поправках к Управлению по контролю за продуктами и лекарствами и постмаркетинговые обязательства». «JAMA» 310 (2): 202–204 doi : 10.1001 / jama.2013.7900.
- Мэдден, Бартли (2010) «Свобода выбора лекарства: как более быстрый доступ к новым лекарствам спасет бесчисленное множество» Жить и положить конец ненужным страданиям Чикаго: Институт Хартленда. ISBN 978-1-934791-32-5
- Мур, Томас Дж. (1998). Рецепт на случай катастрофы: скрытые опасности в вашей аптечке. Нью-Йорк: Саймон и Шустер. ISBN 0-684-82998-3
- Обенчейн, Джанель и Арлин Спарк. Продовольственная политика: взгляд в прошлое. CRC Press, 2015.
Внешние ссылки
| Викискладе есть медиафайлы, связанные с Управлением по контролю за продуктами и лекарствами (США) . |
| Викиновости есть новости, связанные с: Управлением по контролю за продуктами и лекарствами |