Руководство по инструментальным методам исследований при разработке и экспертизе качества лекарственных препаратов

Название: Руководство по инструментальным методам исследований при разработке и экспертизе качества лекарственных препаратов
Автор: Быковский С.Н. (Ред.)
Издательство: Перо
Год: 2014
Cтраниц: 656
Формат: djvu
Размер: 20 мб
Язык: русский
Книга раскрывает многообразие и специфику инструментальных методов анализа как одного из ключевых методов оценки качества лекарственных препаратов. В издании приводится широкий перечень современных высокотехнологичных приборов с предельно четкими характеристиками, которые позволяют обеспечить получение правильных результатов. В данном издании читатели найдут не только теоретические знания, но и познакомятся с практикой применения высокотехнологичных приборов.
Скачать Быковский С.Н. (Ред.) — Руководство по инструментальным методам исследований при разработке и экспертизе качества лекарственных препаратов

- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Свечкарь В.П.
1
Буданова Н.А
1
Григорьева И.В.
2
Пирогова И.М.
1
1 ГБУ РО «Центр по сертификации и контролю качества лекарственных средств»
2 ГБОУ ВПО РязГМУ Минздрава России
Представлено понятие валидации и ее метрологических характеристик, приведены формулы расчета прецизионности и описана возможность использования методов валидации в фармацевтической практике на примере анализа примесей в спирте этиловом методом газовой хроматографии. Приведены и обобщены результаты анализа спирта этилового на содержание в нем спирта метилового и других токсических микропримесей (пропанола-2) в условиях Испытательной контрольно-аналитической лаборатории ГБУ РО «Центр по сертификации и контролю качества лекарственных средств». Пригодность метода для оценки качества спирта этилового 95 % подтверждена характеристикой «прецизионность», которая включает два показателя – повторяемость и воспроизводимость. Результатом проведенных анализов является разработка Стандартной операционной процедуры «Валидация газохроматографического метода испытаний спирта этилового 95 %».
валидация
прецизионность
повторяемость
воспроизводимость
этиловый спирт
контроль качества
1. Руководство по инструментальным методам исследований при разработке и экспертизе качества лекарственных препаратов / под ред. Быковского С.Н. проф. д.х.н. Василенко И.А., к.м.н. Харченко М.И., к.фарм.н. Белова А.Б., к.фарм.н. Шохина И.Е., к.п.н. Дориной Е.А. – М., Изд-во «Перо», 2014. – С. 27–124, 588–600.
2. ГОСТ Р 51698-2000, изменение № 1 к ГОСТ Р 51698-2000 Водка и спирт этиловый. Газохроматографический метод определения содержания токсичных микропримесей. – М., 2005.
3. ГОСТ Р ИСО 5725-2002 ч. 1–6 Точность (правильность и прецизионность) методов и результатов измерений. – М., 2009.
4. ГОСТ Р ИСО 9000-2008. Системы менеджмента качества. Основные положения и словарь. ISO 9000:2005 Quality management systems – Fundamentals and vocabulary. – М.: Стандартинформ, 2009.
5. Руководство Правила надлежащего производства лекарственных средств для медицинского применения и для ветеринарного применения Таможенного союза (правила надлежащей производственной практики – Good Manufacturing Practice – GMP) Режим доступа:. http://www.aipm.org/upfile/doc/AIPM-GMP_Feb-01-2013.pdf.
6. Фармацевтическая разработка: концепция и практические рекомендации. Научно-практическое руководство для фармацевтической отрасли / под ред. Быковского С.Н. проф. д.х.н. Василенко И.А., проф. к.фарм.н. Деминой Н.Б., к.фарм.н. Шохина И.Е. ,к.х.н. Новожилова О.В., Мешковского А.П., Спицкого О.Р. – М., Изд-во «Перо», 2014. – С. 75–82.
Валидация в соответствии с Правилами GMP – это действия, которые доказывают, что определенная методика, процесс, оборудование, сырье, деятельность или система действительно приводят к ожидаемым результатам [5].
Согласно российского стандарта ГОСТ Р ИСО 9000-2008, понятие валидация определена как подтверждение посредством представления объективных свидетельств того, что требования, предназначенные для конкретного использования или применения, выполнены [4].
Основой для проведения процедуры валидации служат методы статистической обработки результатов анализа, достаточно популярно изложенные применительно к области фармации в ряде публикаций [1, 6], на которые и опираются авторы данной статьи.
Валидация метода контроля качества – это процесс установления характеристик метода, показателей его эффективности и определения его ограничений [6]. Главной задачей валидации аналитической методики является экспериментальное доказательство того, что данная методика пригодна для достижения тех целей, для которых она предназначена. При валидации аналитических методов в фармации в основном используют такие метрологические характеристики, как прецизионность, точность, воспроизводимость, повторяемость, правильность, чувствительность, устойчивость, линейность.
Основной характеристикой аналитических методик для количественного определения лекарственных средств и количественной оценки примесей является прецизионность.
Прецизионность является общим термином для выражения изменчивости повторяющихся измерений. Согласно ГОСТ прецизионность – это степень близости друг к другу независимых результатов измерений, полученных в конкретных регламентированных условиях [3].
Прецизионность аналитической процедуры выражает близость значений (степень разброса) между сериями измерений, полученных в результате многократного анализа одного и того же образца при заданных условиях.
Она выражается как коэффициент вариации в % (относительное стандартное отклонение, %RSD) для статистически значимого количества образцов (n ≥ 10).
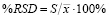 , (1)
, (1)
где S – среднеквадратическое отклонение экспериментальных величин,
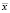 – концентрация анализируемого вещества (среднее значение).
– концентрация анализируемого вещества (среднее значение).
Среднеквадратичное отклонение от среднего (стандартное отклонение выборки, S) является мерой рассеяния (дисперсии) результатов измерений и рассчитывается по формуле (2):
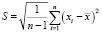 , (2)
, (2)
где xi – результат эксперимента.
В измерениях принято использовать несколько типов прецизионности, в частности таких, как: повторяемость (сходимости, r) и воспроизводимость (R) результатов.
Повторяемость выражает прецизионность при одинаковых рабочих условиях на протяжении короткого промежутка времени, то есть является мерой прецизионности при выполнении ряда условий:
– работает один и тот же исследователь,
– используется один и тот же прибор,
– используется один и тот же метод,
– анализы выполняются в течении короткого промежутка времени,
– используется идентичный объект испытания,
– исследования проводятся в одной и той же лаборатории.
Мерой повторяемости является стандартное отклонение повторяемости (Sr ) и предел повторяемости (r). Предел повторяемости (сходимости) – это значение, которое с доверительной вероятностью 95 % не превышается абсолютной величиной разности между результатами двух измерений (или испытаний), полученными в условиях повторяемости (сходимости).
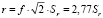 , (3)
, (3)
где f – коэффициент критического диапазона.
Величина f (коэффициент критического диапазона) зависит от доверительного уровня вероятности и закона распределения случайной величины. Для пределов воспроизводимости и повторяемости доверительный уровень вероятности составляет 95 %, и в ГОСТ Р ИСО 5725 делается допущение, что лежащее в основе распределение является приближенно нормальным. Для нормального распределения на уровне вероятности 95 % коэффициент равен 1,96 [3].
Воспроизводимость результатов выражает прецизионность, оцененную по результатам, полученным разными исследователями (внутрилабораторная) или в разных лабораториях (межлабораторная) при выполнении ряда условий:
– работают разные исследователи,
– используется один и тот же метод,
– используется идентичный объект испытания,
– исследования проводятся в одной и той же (внутрилабораторная) или разных лабораториях.
При внутрилабораторных испытаниях прецизионности наблюдения осуществляются в одной и той же лаборатории, но при этом один или несколько факторов – «время», «оператор» или «оборудование» – могут меняться. При установлении прецизионности метода измерений очень важно точно определить соответствующие условия наблюдения, т.е. должны ли быть три вышеупомянутых фактора неизменными или нет. Внутрилабораторные испытания в различные моменты времени учитывают влияние изменения условий окружающей среды и перекалибровки оборудования между наблюдениями. Если в условиях повторяемости наблюдения осуществляются при неизменности всех внутрилабораторных факторов, то в условиях воспроизводимости эти факторы наоборот изменчивы. Если наблюдения выполняются в различных лабораториях, проявляются дополнительные эффекты, являющиеся следствием различия между лабораториями (в административном управлении, материально-техническом обеспечении, проверке стабильности наблюдений и т.д.).
Мерой воспроизводимости является стандартное отклонение воспроизводимости (SR ) и предел воспроизводимости (R = 2,77 SR).
Необходимость рассмотрения прецизионности по ГОСТ Р ИСО 5725-2002 [3] возникает из-за того, что измерения, выполняемые на предположительно идентичных материалах, при предположительно идентичных обстоятельствах, не дают, как правило, идентичных результатов. Это объясняется неизбежными случайными погрешностями, присущими каждой измерительной процедуре, а факторы, оказывающие влияние на результат измерения, не поддаются полному контролю. При практической интерпретации результатов измерений эта изменчивость должна учитываться. Различия между результатами измерений, выполняемых разными операторами и/или с использованием различного оборудования, как правило, будут больше, чем между результатами измерений, выполняемых в течение короткого интервала времени одним оператором с использованием одного и того же оборудования.
Как применяются методы валидации в фармацевтическом анализе, можно проанализировать на примере анализа спирта этилового, который осуществляется в соответствии с ГОСТ Р 51698-2000, изм.1 «Газохроматографический экспресс-метод определения содержания токсичных микропримесей» [2].
Целью исследования является:
– доказательство пригодности метода для оценки качества лекарственных средств в соответствии с требованиями нормативных документов в условиях Испытательной контрольно-аналитической лаборатории ГУЗ «Центр сертификации и контроля качества лекарственных средств Рязанской области»;
– определение характеристики прецизионность, которая включает два показателя: повторяемость и воспроизводимость;
– разработка Стандартной операционной процедуры (СОП) «Валидация газохроматографического метода испытаний спирта этилового 95 %».
Газохроматографический метод применяется для анализа летучих веществ, либо веществ, которые могут быть переведены в парообразное состояние. В газовом хроматографе происходит разделение токсических микропримесей, содержащихся в спирте этиловом и последующее их детектирование пламенно-ионизационным детектором. В нашем случае применяется метод абсолютной градуировки, основанный на предварительном определении зависимости между количеством введенного вещества и площадью пика на хроматограмме. Полученная хроматограмма служит основой для качественного и количественного анализа токсических микропримесей в спирте этиловом.
Определение показателя «Повторяемость» выполняется одним провизором-аналитиком путем анализа 10 проб одного и того же образца трех серий спирта этилового 95 %.
Определение показателя «Воспроизводимость» оценивается по результатам анализов, выполненными параллельно двумя провизорами-аналитиками путем анализа 10 проб одного и того же образца трех серий спирта этилового 95 %.
Для примера приведены результаты испытания одной серии спирта этилового одним аналитиком по количественному содержанию метанола (табл. 1) и пропанола-2 (табл. 2) в анализируемом образце. По результатам испытаний проведён расчет среднеквадратичного отклонения (Sr) и коэффициента вариации ( %RSD). Результаты остальных испытаний по показателю повторяемости сведены в табл. 3. Результаты испытаний по показателю воспроизводимости сведены в табл. 4, по результатам испытаний проведён расчет среднеквадратичного отклонения (SR) и коэффициента вариации ( %RSD).
Таблица 1
Результаты анализа одного образца этилового спирта 95 % на содержание метанола, проведенные одним провизором-аналитиком
|
№ анализа |
Содержание метанола, об. %, xi |
|
|
|
1 |
1,8275*10-3 |
– 0,0395*10-3 |
0,00156*10-6 |
|
2 |
1,8444*10-3 |
0,0226*10-3 |
0,000511*10-6 |
|
3 |
1,7779*10-3 |
0,089*10-3 |
0,007921*10-6 |
|
4 |
1,9315*10-3 |
– 0,0645*10-3 |
0,00416*10-6 |
|
5 |
1,8158*10-3 |
0,0512*10-3 |
0,002621*10-6 |
|
6 |
1,9028*10-3 |
– 0,0358*10-3 |
0,001282*10-6 |
|
7 |
1,986*10-3 |
– 0,119*10-3 |
0,014161*10-6 |
|
8 |
1,8702*10-3 |
– 0,0032*10-3 |
0,00001*10-6 |
|
9 |
1,8275*10-3 |
0,0395*10-3 |
0,00156*10-6 |
|
10 |
1,8670*10-3 |
– 0,019*10-3 |
0,000361*10-6 |
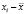
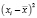
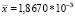
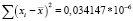
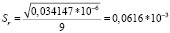
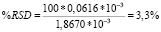
Заключение: коэффициент вариации ( %RSD) для 10 измерений по метанолу составил 3,3 %, что соответствует требованиям ГОСТ (не более 5 %) [3].
Таблица 2
Результаты анализа одного образца этилового спирта 95 % на содержание пропанола-2, проведенные одним провизором-аналитиком
|
№ анализа |
Содержание пропанола-2, мг/дм3, xi |
|
|
|
1 |
2,88*10-4 |
0,12*10-4 |
0,014*10-8 |
|
2 |
2,82*10-4 |
0,06*10-4 |
0,003*10-8 |
|
3 |
2,68*10-4 |
– 0,085*10-4 |
0,007*10-8 |
|
4 |
2,6*10-4 |
– 0,16*10-4 |
0,025*10-8 |
|
5 |
2,77*10-4 |
0,008*10-4 |
0,00006*10-8 |
|
6 |
2,8*10-4 |
0,066*10-4 |
0,004*10-8 |
|
7 |
2,79*10-4 |
0,028*10-4 |
0,00008*10-8 |
|
8 |
2,84*10-4 |
0,077*10-4 |
0,006*10-8 |
|
9 |
2,66*10-4 |
0,097*10-4 |
0,009*10-8 |
|
10 |
2,75*10-4 |
0,01*10-4 |
0,00009*10-8 |
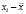
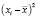
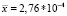
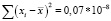
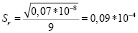

Заключение: коэффициент вариации (RSD) для 10 измерений по содержанию пропанола-2 составил 11,6 %, что соответствует требованиям ГОСТ (не более 5 %) [3].
Таблица 3
Результаты расчета по показателю «повторяемость» (сходимость) испытаний по каждой из серий спирта этилового 95 % по содержанию метанола и пропанола-2
|
условный № серии |
№ аналитика |
%RSD по содержанию метилового спирта |
%RSD по содержанию других токсических микропримесей |
|
1 |
1 |
3,3 |
4,6 |
|
2 |
1 |
4,9 |
5,0 |
|
3 |
1 |
4,8 |
2,9 |
|
1 |
2 |
4,7 |
4,2 |
|
2 |
2 |
3,5 |
4,8 |
|
3 |
2 |
2,4 |
3,2 |
Заключение: коэффициенты вариации (RSD) по метиловому спирту и по содержанию других токсических микропримесей соответствует ГОСТ (не более 15 %) [3].
Аналогично произведен расчет по содержанию пропанола-2, коэффициент вариации ( %RSD) составил 5,79 % что соответствует требованию ГОСТ – должен быть не более 7 % [3].
Из результатов испытаний, проведенных двумя аналитиками в одной и той же лаборатории на одном и том же приборе, следует, что показатели повторяемости (сходимости) и воспроизводимости не превышают указанные пределы, установленные ГОСТ.
Повторная валидация обычно проводится при изменении условий проведения метода и по истечении определенного промежутка времени, в данном случае организация установила срок проведения повторной плановой валидации через пять лет.
Таблица 4
Результаты расчета по показателю «воспроизводимость» по одной из серий спирта этилового 95 % по содержанию метанола
|
Испытатель |
Содержание спирта метилового, %
(среднее из 10 анализов), |
|
|
|
Аналитик № 1 |
1,867*10-3 |
– 0,005*10-3 |
0,000025*10-6 |
|
Аналитик № 2 |
1,877*10-3 |
0,005*10-3 |
0,000025*10-6 |







Заключение: коэффициент вариации ( %RSD) составил по метанолу
0,38 % , что соответствует требованию ГОСТ – должен быть не более 6 %
[3].
Выводы
Пригодность метода для оценки качества спирта этилового 95 % в соответствии с требованиями нормативных документов (ОФС) в условиях Испытательной контрольно-аналитической лаборатории ГУЗ «Центр сертификации и контроля качества лекарственных средств Рязанской области» подтверждена характеристикой «Прецизионность», которая включает два показателя – повторяемость и воспроизводимость. Разработана Стандартная операционная процедура (СОП) «Валидация газожидкостного метода испытаний спирта этилового 95 %», обеспечивющая надежность результатов и пригодная для целей определения качества спирта этилового.
Библиографическая ссылка
Свечкарь В.П., Буданова Н.А, Григорьева И.В., Пирогова И.М. ИСПОЛЬЗОВАНИЕ ВАЛИДАЦИИ В ФАРМАЦЕВТИЧЕСКОЙ ПРАКТИКЕ НА ПРИМЕРЕ ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ В СПИРТЕ ЭТИЛОВОМ // Международный журнал прикладных и фундаментальных исследований. – 2015. – № 7-2.
– С. 263-267;
URL: https://applied-research.ru/ru/article/view?id=7006 (дата обращения: 25.04.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
РАЗРАБОТКА И ВАЛИДАЦИЯ МЕТОДИКИ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДЕСМЕТИЛСИБУТРАМИНА МЕТОДОМ ГЖХ С ПЛАМЕННО-ИОНИЗАЦИОННЫМ ДЕТЕКТИРОВАНИЕМ
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Стерн К.И.
1
Малкова Т.Л.
1
1 ГБОУ ВПО «Пермская государственная фармацевтическая академия» Минздрава России
Статья посвящена проблеме неконтролируемого употребления сильнодействующих веществ (сибутрамина и его структурных аналогов, метаболитов десметилсибутрамина и дидесметилсибутрамина) в биологически активных добавках к пище. В научной литературе отсутствует какая-либо информация по контролю БАД на наличие сильнодействующих веществ, в то время как в практике судебно-химических исследований имеются факты обнаружения данных веществ в данных продуктах. В связи с этим возникла необходимость усовершенствования процедуры обнаружения сильнодействующих веществ методами качественного и количественного анализа. Разработана методика определения десметилсибутрамина методом газо-жидкостной хроматографии с пламенно-ионизационным детектированием. Аналитическая область методики составила 10 – 1000 мкг/мл. Методика валидирована по показателям линейность, правильность, прецизионность и обладает хорошей воспроизводимостью. Данная методика может использоваться в соответствующих аналитических лабораториях.
валидация
психоактивные вещества
десметилсибутрамин
количественный анализ
газо-жидкостная хроматография
1. Валидация методов контроля качества // Руководство по инструментальным методам исследований при разработке и экспертизе качества лекарственных препаратов / под ред. Быковского С. Н. и др. – М. : Перо, 2014. – Разд. 3.3. – С. 80-116.
2. Единые санитарно-эпидемиологические и гигиенические требования к товарам, подлежащим санитарно-эпидемиологическому надзору (контролю) : [Электронный ресурс] // Евразийская экономическая комиссия. – Режим доступа : http://www.tsouz.ru/KTS/KTS17/Pages/P2_299.aspx. – Загл. с экрана (дата обращения: 24.07.14).
3. Об утверждении списков сильнодействующих и ядовитых веществ для целей статьи 234 и других статей Уголовного кодекса Рос. Федерации, а также крупного размера сильнодействующих веществ для целей статьи 234 Уголовного кодекса Рос. Федерации [Электронный ресурс]: постановление Правительства Российской Федерации от 29 декабря 2007 года № 964 // Информационно-правовой портал Гарант. – Режим доступа : http://base.garant.ru/12158202/. – Загл. с экрана (дата обращения: 21.10.14).
4. Сыромятников, С. В. Криминалистическое исследование препаратов, содержащих сибутрамин / С. В. Сыромятников, И. И. Сарычев, Е. А. Гайдукова // Судебная экспертиза. – 2008. — № 4. – С. 61-67.
5. Brain serotonin transporter occupancy by oral sibutramine dosed to teady state: a pet study using 11C-DASB in healthy humans / P. S. Talbot еt al.// Neuropsychopharmacology. – 2009. – Р. 1-11.
6. BTS 54525, A Monoamine uptake inhibitor exhibiting potent actions in models predictive of potential antidepressant activity / W. R. Buckett et al. // British Journal of Pharmacology. – 1987. — № 90. – Р. 94.
7. Identification of N-desmethylsibutramine as a new ingredient in Chinese herbal dietary supplements / Blachut D. et al. // Problems of Forensic Sciences. – 2007. – LXX. – P. 225-235.
8. Simultaneous determination of sibutramine and N-di-desmethylsibutramine in dietary supplements for weight control by HPLC-ESI-MS / Ziqiang Huang et al. // Journal of Chromatographic Science. – 2008. — № 46. – P. 707-711.
Проблема злоупотребления психоактивными веществами в России из года в год становится все более актуальной. В последнее время участились случаи обнаружения сибутрамина и его активных метаболитов (М1 и М2) в биологически активных добавках для похудения (БАД). Федеральная Служба РФ по контролю за оборотом наркотиков в Методических рекомендациях по криминалистическому исследованию сибутрамина от 2006 г. сообщала, что, с учетом дофаминовой теории формирования наркологических заболеваний, сибутрамин может вызывать привыкание и зависимость, а клинические описания его действия позволяют предположить использование его потребителями психоактивных веществ в качестве психостимулятора [4]. С 2008 года сибутрамин, а также его структурные аналоги, обладающие схожим психоактивным действием, включены в список сильнодействующих веществ для целей статьи 234 и других статей Уголовного кодекса Российской Федерации [3].
Сибутрамин метаболизируется изоферментом CYP3А4 до деметилированных метаболитов M1 и М2 (моно- и дидесметилсибутрамин), которые обуславливают его терапевтический эффект и являются структурными аналогами сибутрамина [6, 7, 8]. Установлено, что десметилсибутрамин (ДМС) и дидесметилсибутрамин (ДДМС) примерно в 100 раз активнее исходного соединения [5, 8].
Согласно действующему законодательству РФ, сильнодействующие вещества и их аналоги запрещены к использованию в составе БАД [2]. Ввиду сложившейся ситуации вопрос об усовершенствовании процедуры обнаружения посторонних токсикологически важных веществ, не заявленных в составе БАД, стоит достаточно остро. Однако на сегодняшний день в литературных источниках отсутствует какая-либо информация по процедуре контроля БАД на наличие сильнодействующих веществ, в то время как в практике судебно-химических исследований имеются факты обнаружения данных веществ в биологически активных добавках к пище.
Цель исследования
Разработка методики количественного определения активного метаболита сибутрамина, ДМС, методом газо-жидкостной хроматографии (ГЖХ).
Материал и методы исследования
Субстанцию ДМС получали из содержимого капсул «Жуйдэмэн» (500 мг, № 60), содержащих данное вещество, при комнатной температуре следующим образом: содержимое капсул высыпали в чистую стеклянную склянку, залили спиртом этиловым 95% (1:10), встряхивали в течение 15 минут, затем надосадочную жидкость сливали в чистую стеклянную склянку. Оставшийся осадок вновь заливали спиртом этиловым 95% и операции повторяли. Надосадочные жидкости объединяли и выпаривали без нагревания до сухого остатка. Получившийся сухой остаток брался из расчета, что его состав соответствует экстракту содержимого одной капсулы. После перекристаллизации экстракт был проанализирован на газовом хроматографе Agilent 7890A с масс-спектрометром Agilent 5975C, в результате чего была идентифицирована и подтверждена его химическая структура: масс-спектр пика со временем удерживания 13,18 мин соответствовал библиотечному масс-спектру ДМС (рис. 1).
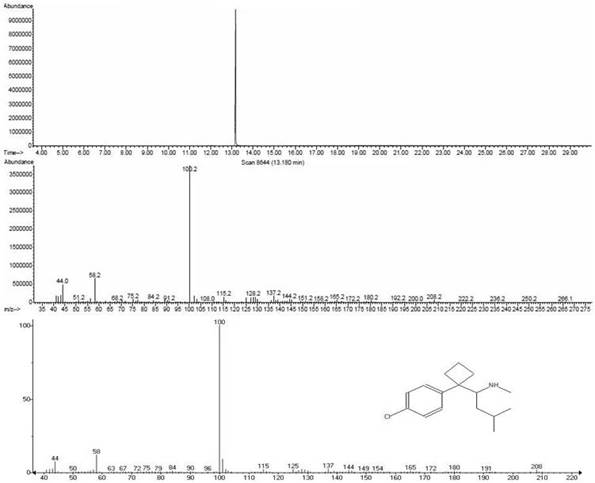
Рисунок 1. Хроматограмма перекристаллизованного экстракта (верхняя часть рисунка), масс-спектр пика с временем удерживания 13,18 мин (средняя часть рисунка) и библиотечный масс-спектр десметилсибутрамина (нижняя часть рисунка).
Готовили растворы ДМС в этиловом 96 % спирте в концентрациях 1 мг/мл, 500 мкг/мл, 200 мкг/мл, 100 мкг/мл, 50 мкг/мл и 10 мкг/мл. В качестве внутреннего стандарта использовали раствор метилстеарата в 96 % этиловом спирте в концентрации 1 мг/мл.
Исследование проводили на газовом хроматографе Хроматэк-Кристалл 5000 с пламенно-ионизационным детектором в следующих условиях: колонка HP-5MS, скорость потока газа-носителя (азот) 2,3 мл/мин, температура термостата колонки начальная 170 °С, конечная – 220 °С, температура детектора 250 °С, температура испарителя 230 °С, ввод пробы с делением потока 1/3, объем вводимой пробы 1 мкл, время хроматографирования 18 мин.
Результаты исследования и их обсуждение
Количественное определение ДМС осуществляли методом ГЖХ с расчетом концентрации по методу внутреннего стандарта, в качестве которого был выбран метилстеарат, преимуществами использования которого являются его хроматографические свойства, близкие к определяемому веществу, стабильность полученных результатов, и доступность для закупки на территории РФ.
Время удерживания ДМС и метилстеарата в заданных условиях хроматографирования составило 7,07 мин и 10,93 мин соответственно (рис. 2).
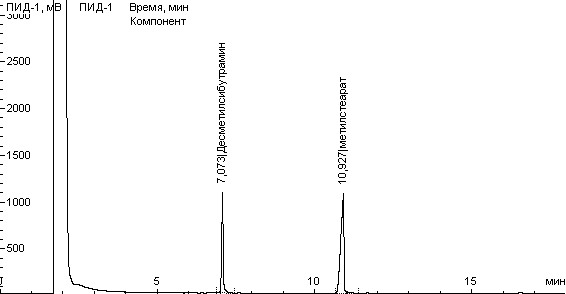
Рисунок 2. Хроматограмма раствора ДМС с внутренним стандартом (метилстеарат) в концентрации 1000 мкг/мл.
Валидацию разработанной методики осуществляли по показателям: линейность, правильность, прецизионность (на уровне intra-day и inter-day), аналитическая область [1].
Линейность
Для определения линейности проводили анализ 6 калибровочных спиртовых растворов ДМС с концентрациями от 10 мкг/мл до 1000 мкг/мл в присутствии внутреннего стандарта (1000 мкг/мл) (табл. 1).
Таблица 1
Значения концентраций калибровочных растворов и отношений площадей пиков ДМС к площадям пиков метилстеарата
|
СДМС, мкг/мл |
СМС, мкг/мл |
SДМС |
SМС |
SДМС/ SМС |
|||
|
Si |
Sср |
Si |
Sср |
Si |
Sср |
||
|
10 |
1000 |
49,919 |
46,512 |
8319,833 |
7752,000 |
0,0060 |
0,0060 |
|
45,663 |
7610,500 |
0,0060 |
|||||
|
43,954 |
7325,667 |
0,0060 |
|||||
|
50 |
1000 |
193,912 |
200,776 |
6002,845 |
6078,020 |
0,0323 |
0,0330 |
|
211,794 |
6310,256 |
0,0336 |
|||||
|
196,621 |
5920,958 |
0,0332 |
|||||
|
100 |
1000 |
303,652 |
405,149 |
5855,909 |
7692,778 |
0,0519 |
0,0526 |
|
489,749 |
9227,440 |
0,0531 |
|||||
|
422,047 |
7994,986 |
0,0528 |
|||||
|
200 |
1000 |
677,061 |
678,460 |
5732,946 |
5754,650 |
0,1181 |
0,1179 |
|
681,875 |
5808,131 |
0,1174 |
|||||
|
676,443 |
5722,872 |
0,1182 |
|||||
|
500 |
1000 |
1550,308 |
1870,566 |
5834,157 |
6914,314 |
0,2660 |
0,2703 |
|
2129,204 |
7808,231 |
0,2730 |
|||||
|
1932,187 |
7100,553 |
0,2720 |
|||||
|
1000 |
1000 |
3814,719 |
3619,324 |
6320,993 |
5998,512 |
0,6035 |
0,6034 |
|
3286,095 |
5446,867 |
0,6033 |
|||||
|
3757,157 |
6227,676 |
0,6033 |
*СДМС – концентрация десметилсибутрамина в растворе, СМС – концентрация метилстеарата в растворе,
SДМС – площадь пика ДМС, SМС – площадь пика метилстеарата.
По полученным значениям строили график линейной зависимости. Были рассчитаны коэффициенты регрессионной прямой у = 0,0006х методом наименьших квадратов, где у – среднее значение отношения площади пика ДМС к площади пика метилстеарата, рассчитанное по трем хроматограммам, х – концентрация ДМС, мкг/мл (Сфакт). (рис. 3).
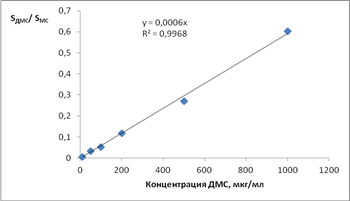
Рисунок 3. Калибровочный график зависимости отношения площади пика ДМС к площади пика метилстеарата от концентрации ДМС в растворе
Квадрат линейного коэффициента корреляции (R2) характеризует степень соответствия между регрессионной моделью и исходными данными. В данном случае 99,68% изменений зависимой переменной описывается регрессионным уравнением. Коэффициент корреляции R = 0,9984, что свидетельствует о наличии прямой линейной зависимости между площадью пика ДМС и его концентрацией в растворе.
Правильность и прецизионность
Для оценки правильности и прецизионности методики проводили анализ 3 калибровочных спиртовых растворов ДМС с концентрациями 10 мкг/мл, 200 мкг/мл, и 1000 мкг/мл в течение первого дня (intra-day) и второго дня (inter-day). Каждый раствор хроматографировали в трех повторностях. Для полученных значений концентрации ДМС рассчитывали величину стандартного отклонения (SD), относительного стандартного отклонения (RSD, %) и отклонение от заданной величины (ε, %). Данные представлены в таблицах 2 и 3.
Таблица 2
Оценка правильности и прецизионности (intra-day)
|
Сфакт, мкг/мл |
Сизм, мкг/мл |
Сср (n=3) |
SD (n=3) |
RSD, % (n=3) |
ε, % |
|
10 |
10,03 |
10,04 |
0,01 |
0,12 |
0,40 |
|
10,03 |
|||||
|
10,05 |
|||||
|
200 |
196,85 |
197,08 |
0,24 |
0,12 |
1,46 |
|
197,33 |
|||||
|
197,07 |
|||||
|
1000 |
1005,83 |
1005,61 |
0,19 |
0,02 |
0,56 |
|
1005,50 |
|||||
|
1005,50 |
*Сфакт – фактическая концентрация ДМС в растворе, Сизм – концентрация ДМС в растворе, рассчитанная по методике.
Таблица 3
Оценка правильности и прецизионности (inter-day)
|
Сфакт, мкг/мл |
Сизм, мкг/мл |
Сср (n=3) |
SD (n=3) |
RSD, % (n=3) |
ε, % |
|
10 |
9,87 |
9,91 |
0,05 |
0,52 |
0,90 |
|
9,90 |
|||||
|
9,97 |
|||||
|
200 |
195,33 |
195,61 |
0,56 |
0,29 |
2,19 |
|
195,25 |
|||||
|
196,25 |
|||||
|
1000 |
1007,83 |
1007,81 |
0,28 |
0,03 |
0,78 |
|
1007,52 |
|||||
|
1008,08 |
*Сфакт – фактическая концентрация ДМС в растворе, Сизм – концентрация ДМС в растворе, рассчитанная по методике.
Полученные значения RSD и ε свидетельствуют о достаточной степени соответствия между истинным значением определяемого вещества и его значением, рассчитанным по данной методике.
Аналитическая область методики
Аналитическая область методики на основании результатов оценки линейности, правильности и прецизионности составила 10 – 1000 мкг/мл.
Заключение
Разработана методика количественного определения десметилсибутрамина методом газовой хроматографии с пламенно-ионизационной детекцией, обладающая необходимой линейностью, правильностью и прецизионностью, что позволяет говорить о хороших валидационных характеристиках данной методики.
Рецензенты:
Вихарева Е.В., д.фарм.н., доцент, заведующий кафедрой аналитической химии ГБОУ ВПО ПГФА Минздрава России, г. Пермь;
Ярыгина Т.И., д.фарм.н., профессор кафедры фармацевтической химии факультета очного обучения ГБОУ ВПО ПГФА Минздрава России, г. Пермь.
Библиографическая ссылка
Стерн К.И., Малкова Т.Л. РАЗРАБОТКА И ВАЛИДАЦИЯ МЕТОДИКИ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДЕСМЕТИЛСИБУТРАМИНА МЕТОДОМ ГЖХ С ПЛАМЕННО-ИОНИЗАЦИОННЫМ ДЕТЕКТИРОВАНИЕМ // Современные проблемы науки и образования. – 2014. – № 6.
;
URL: https://science-education.ru/ru/article/view?id=15483 (дата обращения: 25.04.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)